Exploration of the biodiversity and mining novel target genes of Listeria monocytogenes strains isolated from beef through comparative genomics analysis
Bo Zhang, Wenjie Sun, Xiaoxu Wang, Honglin Ren, Yang Wang, Shaohui Hu, Chengwei Li, Yuzhu Wang, Jiaqi Hou, Xueyu Hu, Ruoran Shi, Yansong Li, Shiying Lu, Qiang Lu, Zengshan Liu, Pan Hu

TL;DR
This study explores the genetic diversity of Listeria monocytogenes in beef and identifies new target genes for its detection.
Contribution
The study identifies novel hub genes, including bglF_2, as potential molecular targets for detecting L. monocytogenes in beef.
Findings
L. monocytogenes strains isolated from beef have open genomes and show high genetic adaptability.
The bglF_2 gene demonstrated excellent specificity and detection capability for L. monocytogenes in beef samples.
Ten hub genes were identified as crucial for L. monocytogenes pathogenesis and detection.
Abstract
L. monocytogenes is a significant foodborne pathogen. This study aims to explore the biodiversity and evolutionary characteristics of L. monocytogenes isolated from beef through pan-genome analysis, and to provide important reference value for its specific molecular detection. This study conducted an in-depth analysis of the virulence genes, antimicrobial resistance genes, and environmental resistance genes of 344 L. monocytogenes strains isolated from beef. Pan-genomic analysis revealed that L. monocytogenes from beef have open genomes, providing a solid genetic basis for adaptation to different environments. MLST analysis revealed that the most prevalent types of L. monocytogenes isolated from beef were ST9 and CC9. A total of 50 virulence genes were detected in these strains, with 26 virulence genes such as inlA, inlB, plcA, plcB, and prfA, present in all L. monocytogenes strains.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
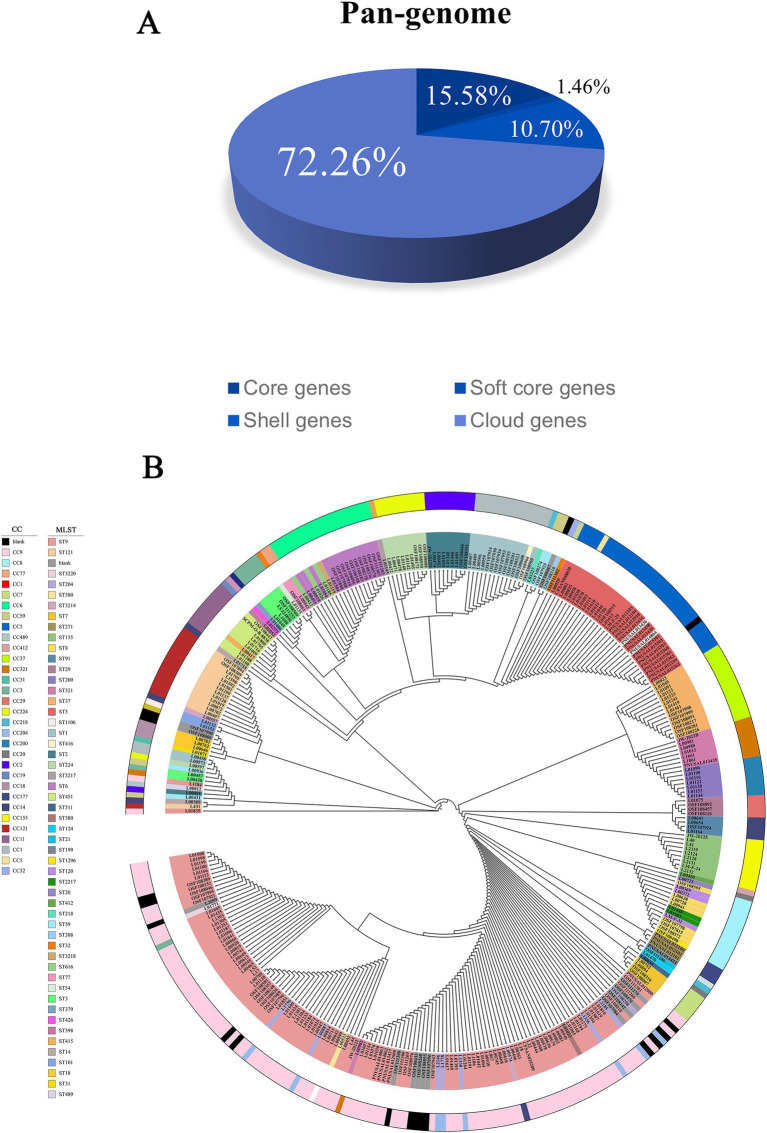 Figure 1
Figure 1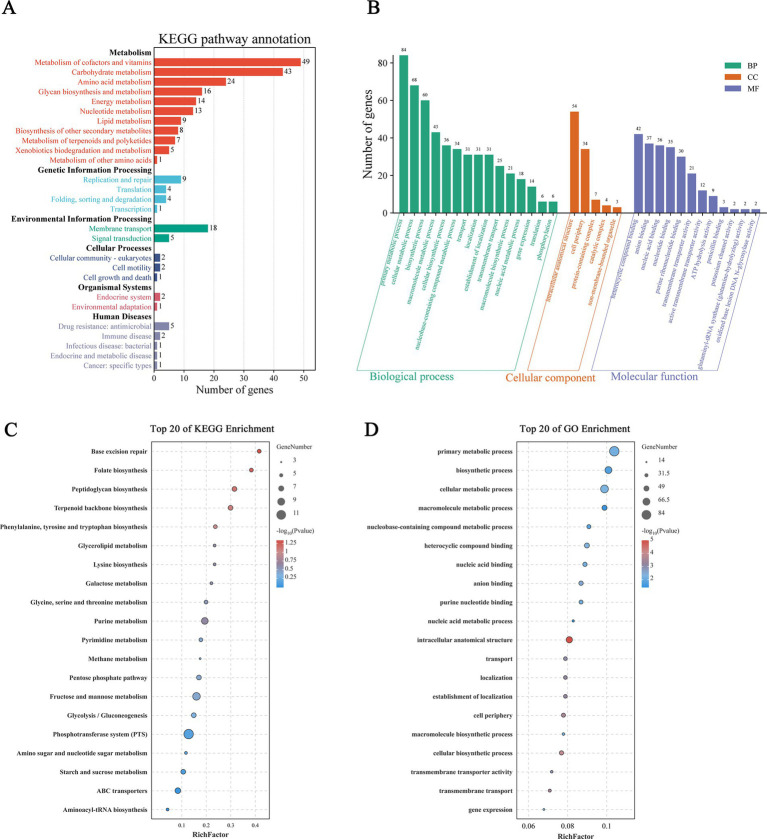 Figure 2
Figure 2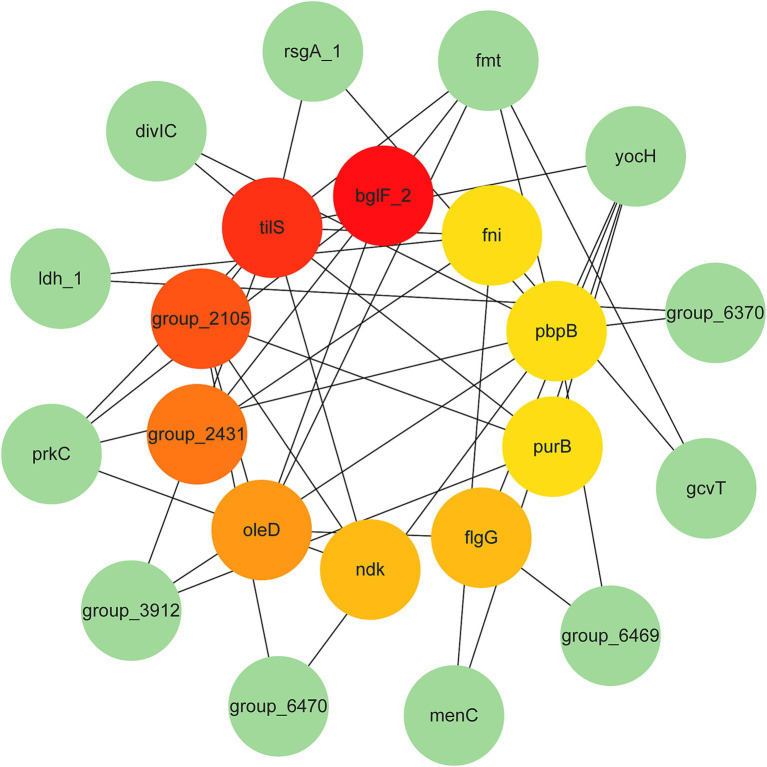 Figure 3
Figure 3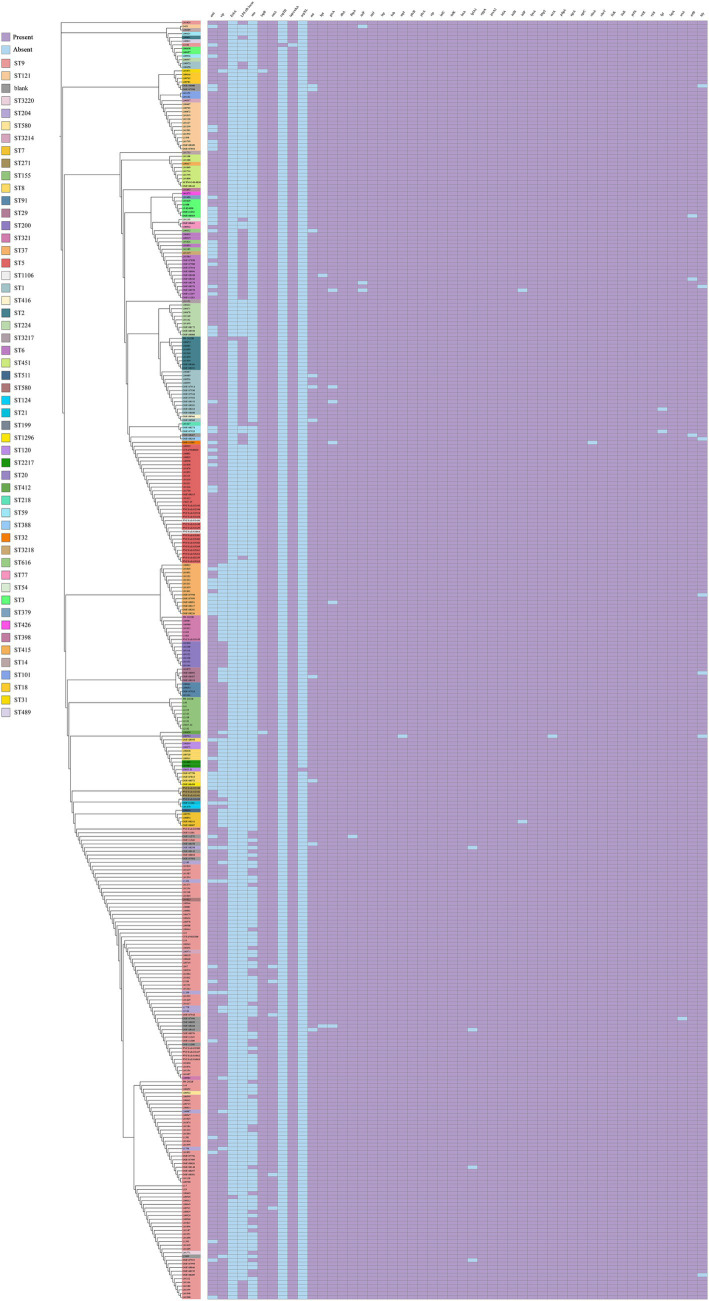 Figure 4
Figure 4 Figure 5
Figure 5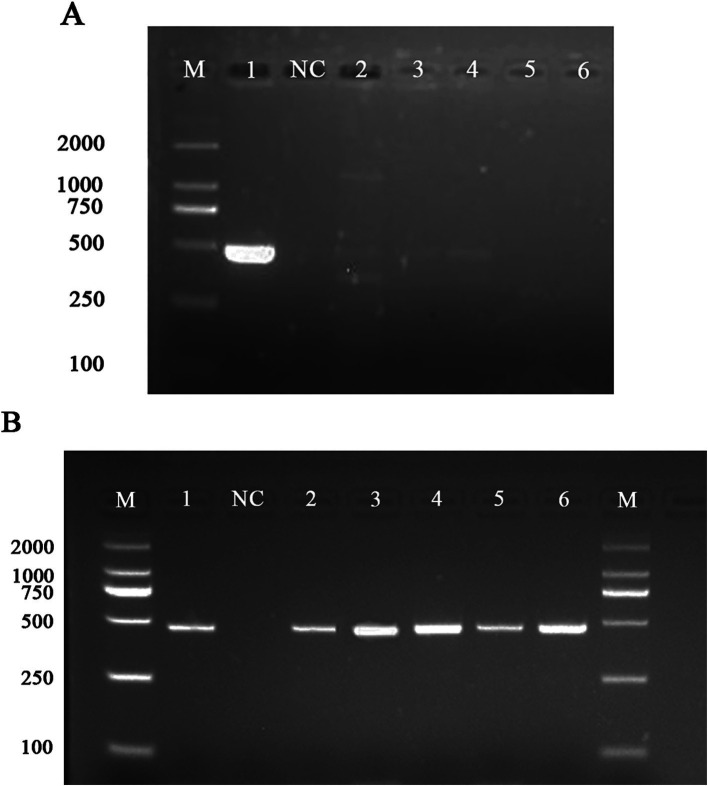 Figure 6
Figure 6| Gene | Sequence length/bp | Primer | Sequence (5′/3′) | Encoded protein | Product size/bp |
|---|---|---|---|---|---|
| bglF_2 | 1,853 | bglF_2F | TCGGAAATGACGTGCCTAAAGTGTT | PTS system beta-glucoside-specific EIIBCA component | 464 |
| bglF_2R | ATCGGAATAACAGAGTAAGC |
| Catelogy | Rank methods in cytoHubba | |||||||
|---|---|---|---|---|---|---|---|---|
| Betweenness | BottleNeck | Closeness | Degree | EPC | MCC | MNC | Stress | |
| Gene symbol top 10 | group_2105 | group_2105 | tilS | bglF_2 | bglF_2 | gatY_3 | bglF_2 | tilS |
| tilS | ndk | group_2105 | tilS | tilS | gatY_2 | tilS | group_2105 | |
| bglF_2 | flgG | bglF_2 | group_2105 | oleD | frwD | group_2431 | bglF_2 | |
| flgG | bglF_2 | oleD | group_2431 | group_2431 | mngA_3 | gatY_3 | ndk | |
| ndk | group_5668 | ndk | oleD | purB | bglF_2 | gatY_2 | flgG | |
| oleD | tilS | group_2431 | ndk | ndk | yhaP | cpoA | oleD | |
| group_2431 | oleD | prkC | flgG | group_2105 | mshD_2 | purB | group_2431 | |
| group_5795 | fmt | pyrE | purB | pyrE | group_10780 | group_11281 | cysG | |
| prkC | hemH | fmt | pbpB | gatY_3 | pgcA_1 | group_5806 | pphA | |
| cysG | cysG | purB | fni | pgcA_1 | pgcA_2 | group_45051 | group_5795 | |
| Gene | Name of target ganes | Sequence length/bp | Presence profile | Encoded protein | Source | |
|---|---|---|---|---|---|---|
| In target | In non-target | |||||
| bglF_2 | lmo2772 | 1,853 | 344 (100%) | 0 | PTS system beta-glucoside-specific EIIBCA component | This study |
| tilS | lmo0219 | 1,945 | 344 (100%) | 0 | tRNA(Ile)-lysidine synthase | This study |
| group_2105 | lmo1266 | 955 | 344 (100%) | 0 | O-succinylbenzoic acid--CoA ligase | This study |
| group_2431 | lmo0078 | 1,350 | 344 (100%) | 0 | Endonuclease MutS2 | This study |
| oleD | lmo1477 | 1,027 | 344 (100%) | 0 | Putative N-acetyl-LL-diaminopimelate aminotransferase | This study |
| ndk | ndk | 443 | 344 (100%) | 0 | 5-dehydro-2-deoxygluconokinase | This study |
| flgG | lmo0682 | 779 | 344 (100%) | 0 | Glutathione biosynthesis bifunctional protein GshAB | This study |
| purB | purB | 1,292 | 344 (100%) | 0 | Siroheme synthase | This study |
| pbpB | pbpB | 2,216 | 344 (100%) | 0 | Tryptophan synthase alpha chain | This study |
| fni | lmo1383 | 1,073 | 344 (100%) | 0 | Histidinol-phosphate aminotransferase | This study |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsListeria monocytogenes in Food Safety · Microbial Metabolism and Applications · Identification and Quantification in Food
Introduction
L. monocytogenes is a foodborne pathogen belonging to Gram-positive bacteria. This highly adaptable bacterium can thrive under adverse conditions, including low temperatures, high salinity, and extreme pH levels. It is commonly associated with food contamination, particularly in meat products (Zhang et al., 2021; Montero et al., 2015). It can cause listeriosis in humans, particularly in populations with compromised immune function, such as neonates, the elderly, pregnant women, and individuals with weakened immune systems. The onset of the disease may be accompanied by various symptoms, including mild diarrhea, meningitis, and septicemia (Matle et al., 2020; Colomba et al., 2020). Listeriosis is typically caused by the consumption of contaminated food, making food safety particularly important for the prevention and control of the disease.
The detection of L. monocytogenes in food typically involves biochemical and molecular methods (Gupta and Adhikari, 2022; Law et al., 2015). Biochemical methods are conventional and accurate, but require a 7-day bacterial incubation period followed by morphological, biochemical, and serological confirmation. This approach is labor-intensive and time-consuming, making it unsuitable for rapid detection (Sloan et al., 2017). Molecular methods, on the other hand, can shorten the detection time to a few hours, allowing for precise and rapid identification of the bacteria (Buszewski et al., 2017; Feng et al., 2020). Molecular techniques developed for L. monocytogenes detection include PCR, quantitative PCR (qPCR), and multiplex PCR (Liu, 2013). The hly gene is an important virulence factor in the infection process of L. monocytogenes. It encodes a toxin called listeriolysin O (LLO), which facilitates the escape and infection of the host by L. monocytogenes. As a result, the hly gene has been widely used in PCR-based detection for identifying L. monocytogenes (Le et al., 2011). However, it is important to note that not all L. monocytogenes strains carry the hly gene (Dapgh and Salem, 2022; Ata et al., 2017; Li et al., 2021), indicating the need to mining more specific genes for the detection of L. monocytogenes.
In recent years, with the widespread application of next-generation sequencing and third-generation sequencing technologies, a large number of L. monocytogenes genomes have been sequenced and shared (Lakicevic et al., 2023; Fagerlund et al., 2022; Fox et al., 2016). This study aims to explore the biodiversity and evolutionary characteristics of L. monocytogenes isolated from beef through pan-genome analysis, and to provide important reference value for its specific molecular detection. Therefore, we conducted a comparative genomics study on all L. monocytogenes strains isolated from beef in the NCBI database. A pan-genomic analysis and Multilocus Sequence Type (MLST) phenotypic analysis were performed for each L. monocytogenes strain. Pan-genomic analysis enables the identification of potential target genes in the strains. Functional analysis of the potential target genes in L. monocytogenes was conducted using Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) annotations. Furthermore, a protein–protein interaction (PPI) network was constructed for potential target genes of L. monocytogenes, and eight different hub gene analysis methods were utilized to screen novel target genes from the potential target genes. Finally, the virulence genes, antimicrobial resistance genes, CRISPR-Cas system, plasmids, and environmental resistance genes of each L. monocytogenes strain were investigated to explore their biodiversity and genetic determinants.
Materials and methods
Data availability and processing
A total of 356 genomic sequences were retrieved and downloaded from the NCBI Genomic Database (last accessed on July 13, 2024), including 344 L. monocytogenes strains isolated from beef, as well as genomic sequences from 5 other Listeria species and 7 non-Listeria bacterial species. The comparative genomic analysis of L. monocytogenes with 5 other Listeria species effectively distinguishes L. monocytogenes from other Listeria species. Similarly, the comparative genomic analysis with 7 non-Listeria bacterial species allows for effective differentiation of L. monocytogenes from non-Listeria bacteria. The concrete information of L. monocytogenes in research is summarized in Supplementary Tables S1, S2, including GenBank accession numbers, strain names, genome size, GC content, number of contigs and N50.
Pan-genomics analysis of Listeria monocytogenes strains isolated from beef and non-targeted strains
The analysis of pan-genomic comparison of L. monocytogenes and non-target strains can be used to screen potential target genes. The potential target genes refer to those genes that are unique to L. monocytogenes strains and are absent in non-target strains (Li et al., 2021; Zhang et al., 2024). Our study found that the commonly used molecular detection target gene, hly, is not present in all strains of L. monocytogenes. Therefore, it is essential to identify potential target genes that are present in all strains of L. monocytogenes as new detection targets (Dapgh and Salem, 2022; Ata et al., 2017; Li et al., 2021). Perform pan-genomic analysis on 344 target strains of L. monocytogenes and 12 non-target reference strains. In brief, all analyzed genome sequences were annotated using Prokka v1.14.6 (Seemann, 2014), and the output results of Prokka were used for pan-genomic analysis with Roary v3.11.2 (Page et al., 2015). A core genome was determined for each isolate using a 99% cutoff, with a BLASTP identity cutoff of 85% (Pang et al., 2019). Genes that matched with all L. monocytogenes strains genome sequences were considered highly conserved and used for subsequent comparisons with other Listeria species and non-Listeria bacterial genomes.
Pan-genome clusters were defined as core-genes: present in all isolates; soft-core genes: present in at least 95% of isolates; shell-genes (accessory genes): present between 15 and 95% of isolates; and cloud-genes (unique genes): present in less than 15% of isolates (Mafuna et al., 2022).
The potential target genes were screened according to the following criteria: 100% presence in L. monocytogenes strains and no presence in non-target bacterial strains. Then, these potential target genes were used screened against the nucleotide collection (nr/nt) databases using the online BLAST program to ensure specificity (Zhang et al., 2024).
MLST and phylogenetic analysis
Perform MLST analysis on the genome of L. monocytogenes to predict sequence types (STs), clonal complexes (CCs), and lineages. This analysis involved retrieving seven housekeeping genes from each L. monocytogenes genome using the MLST database: abcZ (ABC transporter), bglA (beta-glucosidase), cat (catalase), dapE (succinyldiaminopimelate desuccinylase), dat (D-alanine aminotransferase), ldh (lactate dehydrogenase), and lhkA (histidine kinase). These genes were employed to determine the STs, CCs, and lineages of L. monocytogenes (Wu et al., 2016; Wang et al., 2021).
To investigate the phylogenetic relationships among 344 strains of L. monocytogenes isolated from beef, MEGA software was utilized to construct a phylogenetic tree based on all core single-copy genes of L. monocytogenes (Li et al., 2021). Additionally, the ST and CC typing of each strain were annotated on the tree.
Functional characteristics of potential target genes
In order to investigate the functional characteristics of genes present exclusively in L. monocytogenes strains and absent in non-target bacterial strains (potential target genes), annotation analysis was performed using GO enrichment analysis and KEGG enrichment analysis (Adnan et al., 2022), and the results were integrated.
PPI network analysis and screening of novel target genes
In this study, the STRING database was used to establish PPI networks, and these networks were visualized using Cytoscape v3.10.2 (Lu et al., 2022). Cytoscape is a general-purpose modeling environment for integrating biomolecular interaction networks and states. The CytoHubba function in Cytoscape was used to identify hub genes (novel target genes) from the PPI network. CytoHubba ranks genes in the PPI network using eight different algorithms, including Degree, Betweenness, BottleNeck, Closeness, Edge Percolated Component (EPC), Maximum Neighborhood Component (MNC), Maximum Clique Centrality (MCC), and Stress. The top 10 genes with the highest scores in each algorithm were selected as hub genes (Zhang et al., 2024).
Prediction of virulence factors and antimicrobial resistance genes of Listeria monocytogenes
Identifying the virulence and antimicrobial resistance genes of L. monocytogenes is crucial for understanding its genetic determinants. Therefore, we used the Virulence Factors of Pathogenic Bacteria (VFDB) database and the Comprehensive Antibiotic Resistance Database (CARD) to predict the virulence and resistance genes of L. monocytogenes strains (Zhu et al., 2023), and integrated the results for presentation using a heatmap.
Prediction of CRISPR-Cas system types and plasmids of Listeria monocytogenes
The presence of the CRISPR-Cas system and plasmids can facilitate the evolution of L. monocytogenes, contributing to our understanding of its genetics. Therefore, we used CRISPRCasFinder and the PLSDB plasmid database to predict the CRISPR-Cas system and plasmids in L. monocytogenes strains (Di et al., 2014; Zhang et al., 2016), and integrated the results for presentation using a heatmap.
Prediction of Listeria genomic Islands and stress survival Islands of Listeria monocytogenes
The presence of Listeria genomic Islands (LGIs) and survival Islands (SSIs) can enhance the resistance of L. monocytogenes to harsh environments, contributing to our understanding of its adaptability. Therefore, we used the BIGSdb database to predict the LGIs and SSIs in L. monocytogenes strains (Mafuna et al., 2021), and integrated the results for presentation using a heatmap.
Design and validation of specific primers for Listeria monocytogenes
Primers for the bglF_2 gene sequence were designed using Primer Premier 5 software (Table 1) (He et al., 2022). The primers were synthesized by Sangon Biotech Co., Ltd., Shanghai, China. PCR experiments on some of the analyzed strains to detect primer specificity. Total reaction volume was 25 μL, including 12.5 μL of 2 × Es Taq MasterMix (CWBIO, Beijing, China), 1 μL each of for ward and reverse primers (10 μM), 8.5 μL of sterile water, and 2 μL of the purified bacterial genomic DNA as a template. An equal volume of sterile distilled water was used instead of the template as a negative control. PCR thermal cycling involved an initial denaturation step at 94°C for 5 min, followed by 35 cycles of denaturation at 94°C for 30 s, annealing at 55°C for 30 s, and elongation at 72°C for 1 min, with a final elongation at 72°C for 10 min. PCR products were evaluated by 2% agarose electrophoresis.
Artificial contamination experiments
To ensure the specificity and efficacy of the bglF_2 gene, the primers designed for this gene were validated through artificial contamination experiments to assess their effectiveness in detecting L. monocytogenes in beef samples. In brief, the L. monocytogenes EGD-e strain was cultured overnight, and bacterial concentration was determined using the plate count method. Beef samples (10 g) obtained from a slaughterhouse in Changchun, China were minced and exposed to ultraviolet light for 30 min to ensure sterility. The samples were then added to 88 mL of Milli-Q water and incubated for 30 min. The mixture was centrifuged at 8,000 rpm for 20 min, and the supernatant was filtered through a 0.22 μm filter to obtain the simulated detection solution. Finally, 2 mL of the cultured EGD-e was added to the simulated detection solution, and the mixture was thoroughly mixed to complete the artificial contamination. After that, the mixtures were incubated at 37°C for 4 h,6 h, 8 h,12 h, and 24 h. Genomic DNA was extracted at the indicated time points from sample, and then analyzed by PCR (Li et al., 2021).
Results
Genome statistics and general features
In the NCBI database, we downloaded the whole-genome sequences of all L. monocytogenes strains isolated from beef, totaling 344 strains, and compiled the corresponding information for these strains (Supplementary Tables S1, S2). They possess an average genome size of 3.061 (2.9–3.6) Mbp, an average GC content of 37.986 (37.5–38)%, the number of contigs ≤ 312, and an average N50 of 387.121 Kbp.
Pan-genomic analysis results
According to the pan-genome analysis, the distribution of pan-genes in 344 L. monocytogenes strains is as follows: 2001 core genes (15.58%), 187 soft core genes (1.46%), 1,374 shell genes (10.70%), and 9,278 cloud genes (72.26%) (Figure 1A). As the number of strains increases, the number of pan-genes gradually increases, while the number of core genes tends to stabilize, indicating that L. monocytogenes possesses an open genome, which is closely related to their adaptability and virulence expression in beef environments under different conditions. To identify potential target genes, we also performed pan-genome analysis on 344 L. monocytogenes strains and 12 non-target strains. The results of the pan-genome analysis are as follows: the number of core genes is 0, soft core genes are 2,098, shell genes are 1,439, cloud genes are 41,690, and the total number of pan-genes is 45,227. Due to the presence of non-target strains, no common core genes were identified. However, potential target genes can be screened by comparing genes present in the non-target strains. Based on the aforementioned screening method, a total of 416 potential target genes were identified in L. monocytogenes strains (Supplementary Table S3). These potential target genes were present in 100% of the L. monocytogenes strains isolated from beef, but absent in non-target strains. These potential target genes may serve as novel target genes for L. monocytogenes strains isolated from beef, but further screening is required to validate these potential target genes.
The pan-genome composition and phylogenetic evolution tree of L. monocytogenes isolated from beef. (A) The proportion of each component in the pan-genome. (B) The phylogenetic tree of L. monocytogenes isolated from beef, with the inner ring color representing different STs and the outer ring color representing different CCs.
MLST and phylogenetic analysis
By uploading the whole-genome sequences of 344 L. monocytogenes strains to the database for comparison, these strains were classified into 52 distinct ST types and 34 different CC types. However, 18 strains did not successfully match any corresponding types in the database. The most common ST types were ST9 (28.20%) and ST5 (8.43%), while the most common CC type was CC9 (30.23%). The strains were divided into two evolutionary lineages: lineage I (30.23%) and lineage II (64.24%) (Figure 1B; Supplementary Table S4).
To elucidate the evolutionary patterns of L. monocytogenes strains isolated from beef, we performed a phylogenetic analysis of L. monocytogenes using the conserved amino acid sequences of all single-copy genes (Figure 1B). Phylogenetic tree analysis revealed that the L. monocytogenes strains isolated from beef share a common ancestor and belong to the main root of the Listeria genus. Most L. monocytogenes strains isolated from the same region clustered into the same branch, suggesting a distinct regional pattern in the evolution of L. monocytogenes. However, some strains, although isolated from different regions, were grouped into the same branch, indicating a certain level of evolutionary similarity and strong adaptability of L. monocytogenes to different environments.
Enrichment analysis of the functional characteristics of potential target genes using the GO and KEGG databases
To investigate the functional characteristics of the 416 potential target genes from L. monocytogenes strains isolated from beef, we performed functional annotation and classification of these genes using the GO and KEGG databases. The detailed information of the potential target genes is presented in Supplementary Table S3. The pathway database of KEGG is the most widely used public database for metabolic pathways, which classifies biological metabolic pathways into six categories: Metabolism, Genetic Information Processing, Environmental Information Processing, Cellular Processes, Organismal Systems, and Human Diseases. The potential target genes were annotated according to six pathway categories in the KEGG database. Among these, the most enriched pathways in the Metabolism category were metabolism of cofactors and vitamins (n = 49, 11.78%), carbohydrate metabolism (n = 43, 15.87%), and amino acid metabolism (n = 24, 5.78%). In the Genetic Information Processing category, replication and repair (n = 9, 2.16%) was the most enriched pathways. In the Environmental Information Processing category, membrane transport (n = 18, 4.33%) and signal transduction (n = 5, 1.20%) were the most enriched pathways. In the Cellular Processes category, cellular community—eukaryotes (n = 2, 0.48%) and cell motility (n = 2, 0.48%) were the most enriched pathways. In the Organismal Systems category, endocrine system (n = 2, 0.48%) was the most enriched pathways. In the Human Diseases category, drug resistance: antimicrobial (n = 5, 1.2%) was the most enriched pathways (Figure 2A). We integrated and ranked the KEGG enrichment analysis results of all potential target genes, and generated bubble maps to show the top 20 functional features in KEGG enrichment analysis based on the significance of gene number and p-value (Figure 2C).
Enrichment analysis of potential target genes of L. monocytogenes isolated from beef based on GO and KEGG databases. (A) Enrichment analysis based on the KEGG database. (B) Enrichment analysis based on the GO database. (C) Enrichment analysis based on KEGG database with the top 20 enriched terms listed. (D) Enrichment analysis based on GO database with the top 20 enriched terms listed.
The GO database categorizes gene functions into three main categories, namely Biological Processes (BP), Cellular Components (CC), and Molecular Functions (MF). In the BP category, the most enriched biological processes were primary metabolic process (n = 84, 20.19%), cellular metabolic process (n = 68, 16.35%), biosynthetic process (n = 60, 14.42%), and macromolecule metabolic process (n = 43, 10.34%). Within the CC category, the most abundant cellular components were intracellular anatomical structure (n = 54, 12.98%), cell periphery (n = 34, 8.17%). In the MF category, the most enriched molecular functions were heterocyclic compound binding (n = 42, 10.09%), and anion binding (n = 37, 8.89%) (Figure 2B). We integrated and ranked all the GO enrichment analysis results of potential target genes, and generated a bubble chart to display the top 20 functional features in GO enrichment analysis based on the number of genes and the significance of p-values (Figure 2D).
Enrichment analysis of the functional characteristics of the 416 potential target genes using the GO and KEGG databases revealed that these genes are primarily associated with metabolic processes, compound binding, protein localization, and transmembrane transport in L. monocytogenes. Specifically, these genes are involved in cellular metabolic processes, carbohydrate metabolism, and biosynthesis of organic matter. For example, carbohydrate metabolism is critical for energy acquisition in L. monocytogenes because it involves converting sugars into a form of energy that cells can use directly. These potential target genes are closely related to the basic biological processes of L. monocytogenes and the pathogenesis of infection. They not only support the basic metabolic activities of the bacteria, but also involve the invasion and pathogenic effects of the bacteria on the host, including how the bacteria survive in the host cell, replicate and evade the innate immune response of the host. However, there were still some genes with unclear functional information, which warrants further investigation in future studies. In conclusion, the functional characteristics of these genes provide deeper insights into the biological properties of L. monocytogenes and offer potential molecular targets for the development of new control strategies.
PPI network analysis of potential target genes and identification of novel target genes
To further explore the relationship between potential target genes of L. monocytogenes PPI analysis was carried out using the STRING database. The PPI network of potential target genes comprised 416 genes, and visualization of the PPI network was performed using Cytoscape v3.10.1 software. Cytoscape is a general-purpose modeling environment for integrating biomolecular interaction networks and states. Potential target genes in the PPI network were screened using eight different algorithms in the CytoHubba plugin of Cytoscape software. The top 10 genes with the highest scores were selected as hub genes, and their rankings are shown in Table 2. Finally, the top 10 hub genes with the highest scores obtained from the Degree algorithm were identified as novel target genes. A PPI network was constructed based on their scores. These 10 hub genes include bglF_2, tilS, group_2105, group_2431, oleD, ndk, flgG, purB, pbpB, and fni (Figure 3). Detailed information on these 10 hub genes, including their functional roles, gene lengths, and other characteristics, is presented in Table 3. These 10 hub genes play a crucial role in maintaining the basic life activities and infection processes of L. monocytogenes strains and may serve as novel target genes for L. monocytogenes, particularly the top-scoring gene, bglF_2.
PPI network analysis of novel target genes of L. monocytogenes isolates from beef. The darker the red, the higher the gene score.
Prediction results of virulence and antimicrobial resistance genes
To investigate the virulence relationships and pathogenic mechanisms among L. monocytogenes strains isolated from beef, we predicted the virulence factor encoding genes in the whole genome of L. monocytogenes strains. Based on VFDB prediction and annotation, virulence factors of L. monocytogenes were classified into 12 categories including Adherence, Bile resistance, Enzyme, Immune modulator, Intracellular survival, Invasion, Iron uptake, Nucleation-promoting factor, Peptidoglycan modification, Regulation, Surface protein anchoring, and Toxin. The VFDB prediction revealed that 26 virulence genes were present in all L. monocytogenes strains isolated from beef, including dltA, inlJ, lap, bsh, plcB, plcA, stp, inlC, inlK, lntA, oppA, prsA2, inlA, inlB, lpeA, hbp2, pdgA, agrA, agrC, cheY, lisK, lisR, prfA, virR, virS, and lspA (Figure 4). During the process of invading and infecting the host, the genes inlA, inlB, plcA, plcB, prfA, hly, and actA play critical roles. In this study, the inlA, inlB, plcA, plcB, and prfA genes were present in 100% of the L. monocytogenes strains, while the hly and actA genes were not present in all strains, but their presence probability exceeded 97%. The prediction of virulence genes indicates that L. monocytogenes strains isolated from beef contain a large number of virulence genes, suggesting that these strains possess high virulence and pathogenicity.
The distribution of virulence genes in L. monocytogenes isolated from beef.
With the widespread use of antibiotics, bacterial resistance has become a major concern. The changes in antimicrobial resistance genes during bacterial evolution help us gain a deeper understanding of the trends in bacterial antimicrobial resistance. Using the CARD database, a total of 9 antimicrobial resistance genes were predicted in 344 L. monocytogenes strains isolated from beef, belonging to 9 drug categories and exhibiting 5 resistance mechanisms. The identified antimicrobial resistance genes include norB, lin, mprF, FosX, dfrG, APH(3′)-IIIa, ANT(6)-Ia, lsaE, and catA8 (Figure 5). The most common 4 antimicrobial resistance genes in L. monocytogenes strains isolated from beef are norB, lin, mprF, and FosX, indicating high resistance to fluoroquinolones, lincosamides, peptides and phosphonic acid antibiotics.
The distribution of antimicrobial resistance genes, CRISPR-Cas system, plasmids, SSIs and LGIs in L. monocytogenes isolated from beef.
Prediction results of CRISPR-Cas system types and plasmids
The CRISPR-Cas system is an adaptive immune system in bacteria that defends against the invasion of foreign genetic material, such as phages and plasmids. It serves as a defense mechanism found in most bacteria to eliminate foreign plasmid or phage DNA. An analysis of the CRISPR-Cas system in 344 L. monocytogenes strains isolated from beef identified 4 types of CRISPR-Cas systems, each exhibiting distinct Cas genes. The CRISPR-Cas system types detected in L. monocytogenes included CAS-TypeIA (2/344), CAS-TypeIB (105/344), CAS-TypeIIA (30/344), and CAS-TypeIIIA (23/344). In total, 39.24% (135/344) of the L. monocytogenes strains contained at least one CRISPR-Cas system, with CAS-TypeIB (30.52%) and CAS-TypeIIA (8.72%) being the most prevalent (Figure 5). CAS-TypeIA was detected only in ST451 and ST2.
Mobile Genetic Elements (MGEs) refer to a class of genetic elements capable of spreading or transferring within a genome, such as plasmids, which can facilitate the evolution of microorganisms. Therefore, the prediction of plasmids can provide further insights into the evolution of L. monocytogenes. We employed PLSDB databases for the detection of plasmids, and the results only documented plasmids with an identity score of 1. A total of 23 plasmids were predicted in L. monocytogenes strains, with the most prevalent plasmids being pMF4545 (10/344) and pCFSAN100570 (10/344), followed by pLM33 (8/344) and pLIS18 (8/344). The plasmids pMF4545 and pCFSAN100570 were exclusively found in ST9 (Figure 5).
Prediction results of LGIs and SSIs
We predicted the presence of genomic LGIs in L. monocytogenes strains and found that LGI-2 was present in all strains. LGI-1 was less common, detected in only 3 L. monocytogenes strains, while LGI-3 was found in 155 strains (Figure 5).
The SSIs associated with resistance genes in Listeria consist of 2 islands: SSI-1 and SSI-2. SSI-1 consists of the genes lmo0444, lmo0445, pva, gadD, and gadT, while SSI-2 is composed of the genes lin0464 and lin0465. SSI-1 is associated with the tolerance of Listeria to environmental factors such as high NaCl concentrations, high bile salt concentrations, and low pH. In contrast, SSI-2 contributes to the survival of strains under alkaline and oxidative stress, enabling them to persist in food processing environments. Initially, the two genes of SSI-2 were thought to be unique to L. innocua, however, studies have found that these genes are also present in L. monocytogenes of the ST121. Prediction of SSIs revealed that 330 strains (95.93%) of L. monocytogenes harbor SSI-1, while 14 strains (4.07%) of L. monocytogenes harbor SSI-2 (Figure 5). It is noteworthy that all 14 L. monocytogenes strains harboring SSI-2 were identified as ST121. Among these 14 L. monocytogenes strains, 12 were isolated from the Netherlands, 1 from Spain, and 1 from Australia.
Detection of Listeria monocytogenes using specific primers by PCR
To validate the specificity of the bglF_2 gene for L. monocytogenes, primers targeting the bglF_2 gene were designed, and its specificity was evaluated through PCR. PCR results showed that in the primer system designed for the bglF_2 gene, a distinct band was observed at 464 bp only in L. monocytogenes, while no bands were detected in other non-L. monocytogenes strains (Figure 6A). The results indicate that the bglF_2 gene exhibits good specificity for L. monocytogenes, and primers designed for the bglF_2 gene can effectively detect L. monocytogenes.
Verification of primer specificity for the bglF_2 gene of L. monocytogenes and artificial contamination experiments. (A) Lane M: DL DNA 2000 marker, lane NC: negative control, and lanes 1–6: represent 6 different strains, including lane 1: Listeria monocytogenes, lane 2: Listeria innocua, lane 3: Listeria ivanovii, lane 4: Listeria welshimeri, lane 5: Escherichia coli, lane 6: Salmonella enterica. (B) Lane M: DL DNA 2000 marker, lane 1: positive control, lane NC: negative control, lane 2: DNA extracted from samples at 4 h, lane 3: DNA extracted from samples at 6 h, lane 4: DNA extracted from samples at 8 h, lane 5: DNA extracted from samples at 12 h, lane 6: DNA extracted from samples at 24 h.
The results of the artificial contamination experiment
To ensure the primers designed for the bglF_2 gene can effectively detect L. monocytogenes in beef samples, their specificity and efficacy were validated through artificial contamination experiments. L. monocytogenes at a concentration of 5.2 × 10^4^ CFU/mL was added to the simulated detection solution and subjected to 24 h of enrichment culture. DNA was extracted from the samples at 4 h, 6 h, 8 h, 12 h, and 24 h, and PCR validation was performed using primers designed for the bglF_2 gene. The results showed distinct bands at 4 h, 6 h, 8 h, 12 h, and 24 h, indicating that the primers designed for the bglF_2 gene can effectively detect L. monocytogenes in beef samples (Figure 6B). The bglF_2 gene demonstrated excellent specificity and holds promise as a specific molecular target for detecting L. monocytogenes strains in beef samples.
Discussion
Food safety is a critical issue in the field of public health, and L. monocytogenes, as a significant foodborne pathogen, often poses serious food safety risks (Chowdhury and Anand, 2023). Therefore, we conducted a comparative genomic analysis of L. monocytogenes strains isolated from beef to explore their phenotypic and genetic evaluations, revealing their biodiversity and evolutionary traits. We also identified potential target genes and mining novel targets. The aim was to gain a deeper understanding of L. monocytogenes strains and provide important reference value for strain-specific molecular detection, thereby reducing food safety issues in public health.
In this study, we conducted a pan-genome comparative analysis of 344 L. monocytogenes strains isolated from beef to investigate strain phenotypes and genetic evaluations, revealing their biodiversity and evolutionary features. To assess the genomic diversity of L. monocytogenes strains isolated from beef, we performed core/pan-genome analysis. The pan-genome represents all the genes present in the strains, while the core genome represents the essential portion necessary for the presence and shared phenotypic features of specific strains, which are critical for maintaining basic survival and infection capability (Lee et al., 2019). Therefore, we conducted an in-depth study of the core genome to identify potential target genes. A total of 2,001 core genes, accounting for 15.58%, were identified among all L. monocytogenes strains isolated from beef in the NCBI database. These genes constitute the fundamental components essential for the survival and development of L. monocytogenes strains. This suggests that despite evolving in different regions and environments, L. monocytogenes strains still share a significant number of common phenotypic features. In addition to the core genes, the pan-genome also contains many accessory genes, which provide the strains with unique traits (Nwaiwu, 2022). As L. monocytogenes strains are found in various environments, they have evolved specific accessory genes to adapt to these distinct environments and counteract environmental pressures. Through pangenomic analysis, we identified the core genes of L. monocytogenes strains isolated from beef, and subsequently used these core genes to determine potential target genes. The selection criterion for potential target genes was that they were present exclusively in L. monocytogenes strains isolated from beef, and absent in non-target strains. This suggests that the potential target genes are essential and unique to L. monocytogenes, playing a crucial role in its basic survival and pathogenic invasion. Therefore, studying the potential target genes of L. monocytogenes can enhance our understanding of its phenotypic and genetic evaluations, and these potential target genes could also serve as specific molecular detection targets for L. monocytogenes.
To investigate the biodiversity and evolutionary characteristics of L. monocytogenes strains isolated from beef, we performed MLST and phylogenetic analyses. MLST analysis is a crucial tool in the classification and epidemiological study of L. monocytogenes, providing important insights into the genetic diversity and transmission routes of L. monocytogenes strains (Stessl et al., 2014). Our MLST analysis of L. monocytogenes strains isolated from beef revealed that ST9 and CC9 were the most prevalent, followed by ST5 and CC5. Similar findings were reported by Henri et al. (2016) who conducted MLST analysis on L. monocytogenes isolated from food, with ST9 and ST121 being the most common. Manqele et al. (2024) performed MLST analysis of L. monocytogenes in beef and beef products and identified ST9, ST204, ST1, and ST5. Our results are consistent with the findings reported in the above-mentioned literature. This indicates that L. monocytogenes strains in beef predominantly belong to ST9 and CC9 types. By constructing a phylogenetic tree, we found that although the L. monocytogenes strains studied were isolated from different regions and environments, some of the strains were still grouped into the same branch. This suggests that while L. monocytogenes strains have evolved unique traits to adapt to the specific environmental pressures they encounter, they still exhibit a certain degree of genetic similarity in their evolutionary characteristics.
We obtained the core genes of L. monocytogenes strains isolated from beef through pan-genomic analysis, and then identified 416 potential target genes by screening these core genes. To explore the functional roles of these potential target genes, we conducted GO and KEGG enrichment analyses (Zhang et al., 2024). The functional annotation results indicated that the potential target genes are mainly associated with the metabolic processes, compound binding, protein localization, and transmembrane transport of L. monocytogenes, which are closely related to the bacterium’s basic life activities and its ability to invade and infect hosts. The potential target genes are promising candidates for specific targeting of L. monocytogenes (Zhang et al., 2024). However, due to the large number of potential target genes, further screening was performed. A PPI network was constructed, and the hub genes were identified using the Cytohubba function in Cytoscape software. Hub genes are the most critical genes in the PPI network and are used to discover novel target genes (Zhang et al., 2024). Ultimately, 10 hub genes were selected from the potential target genes (bglF_2, tilS, group_2105, group_2431, oleD, ndk, flgG, purB, pbpB, and fni). These hub genes play a crucial role in the basic life activities and infection invasiveness of L. monocytogenes. Among them, the bglF_2 gene scored the highest and exhibited stronger connections with other proteins, indicating its potential as a specific molecular target for L. monocytogenes detection.
L. monocytogenes is a recognized pathogenic strain of Listeria (Disson et al., 2021). To further understand the genetic determinants behind the virulence of L. monocytogenes, we predicted the virulence genes of L. monocytogenes strains isolated from beef. During invasion of the host by L. monocytogenes, the bacterium first utilizes the inlA and inlB genes to bind with the E-Cadherin and Met receptors of the host’s eukaryotic cell membrane, respectively, thereby inducing bacterial uptake through receptor-mediated endocytosis. After internalization, the bacterium is encapsulated within a vacuole, and releases the hly, plcA, and plcB genes to mediate vacuole escape. Subsequently, the actA gene is utilized to induce actin polymerization and generate sufficient force for the bacterium to spread from one cell to another (Osek and Wieczorek, 2022). Our predictive results revealed that 50 virulence genes are present in L. monocytogenes strains isolated from beef, with 26 of these virulence genes being consistently present in all strains. These include key virulence genes involved in the infection and invasion process of L. monocytogenes, such as inlA, inlB, plcA, plcB, and prfA genes. Interestingly, hly and actA genes were not present in all strains, although their occurrence exceeded 97%. This discrepancy could be attributed to prediction errors in the database or genetic variations occurring in individual strains under specific environmental conditions. The presence of a large number of virulence genes contributes to the strong pathogenicity of L. monocytogenes, making it a virulent strain. The genetic determinants behind L. monocytogenes virulence are governed by its virulence genes. These results suggest that although some virulence genes may undergo genetic variation during the evolutionary process, critical virulence genes essential for the infection and invasion of the host remain conserved, playing a crucial role in L. monocytogenes pathogenicity.
To further investigate the genetic determinants behind the antibiotic and environmental resistance of L. monocytogenes, we predicted the presence of antimicrobial resistance genes, CRISPR-Cas systems, plasmids, SSIs, and LGIs in L. monocytogenes strains isolated from beef. Antibiotic resistance has long been a concerning issue (Asghar et al., 2024). Our predictions revealed that the most common antibiotic resistance genes in L. monocytogenes are norB, lin, mprF, and FosX, indicating high resistance to fluoroquinolones, lincosamides, peptides, and phosphonic acid antibiotics. The recommended antibiotics for treating L. monocytogenes infections are ampicillin and gentamicin. The CRISPR-Cas system is an important adaptive immune mechanism in bacteria, used to defend against foreign genetic elements such as phages and plasmids (Liu et al., 2024). Our predictions revealed the presence of a significant number of CAS-TypeIB and CAS-TypeIIA in L. monocytogenes strains, suggesting that L. monocytogenes evolves an environment-specific CRISPR-Cas system during its evolutionary process. Predicting the presence of plasmids in L. monocytogenes helps analyze the variations in MGEs and provides further insights into the evolution of the strains. Our prediction of plasmids revealed that, despite L. monocytogenes being exposed to different external environments, there remains a certain degree of similarity in bacterial genome evolution. The prediction of SSI and LGI in L. monocytogenes provides valuable insights into the changes in the environmental resistance of the bacterium (Hein et al., 2011). We found that LGI-2 was present in all strains, whereas LGI-3 was only present in 155 strains, suggesting that the presence of LGI-2 plays a crucial role in the resistance to environmental stress during the evolution of L. monocytogenes. SSI prediction revealed that SSI-1 was present in the majority of L. monocytogenes strains, whereas SSI-2 was found only in the ST121 strains, indicating that the presence of SSI-1 helps L. monocytogenes survive in harsh environments. Therefore, LGI-2 and SSI-1 are a genetic determinant for environmental resistance in the evolution of L. monocytogenes.
To validate the specificity of the bglF_2 gene, primers were designed for the bglF_2 gene, and its specificity was confirmed through PCR. The results indicate that the bglF_2 gene exhibits excellent specificity for L. monocytogenes. The rationale for selecting genes different from those used in previous studies for detecting L. monocytogenes is that the bglF_2 gene yielded the best results in the analysis of L. monocytogenes strains isolated from beef (Zhang et al., 2024). Meanwhile, the bglF_2 gene, as a component of the PTS system, is typically strain-specific, which helps to avoid cross-reactivity with other Listeria species and non-Listeria bacteria. Therefore, the bglF_2 gene exhibits better specificity in these strains, making it a more suitable choice for detecting L. monocytogenes in beef. Subsequently, an artificial contamination experiment was conducted to verify whether the bglF_2 gene could successfully detect L. monocytogenes in beef samples. The results showed that the bglF_2 gene effectively detected L. monocytogenes at five different time points, indicating that bglF_2 not only efficiently detects L. monocytogenes in beef samples but also performs excellently. Therefore, the bglF_2 gene is expected to be a specific molecular target for detecting L. monocytogenes in beef samples.
Conclusion
In conclusion, we conducted a comparative genomics study to explore the phenotypic and genetic evaluations of L. monocytogenes strains isolated from beef, revealing the biodiversity and evolutionary traits of these strains. A large number of virulence genes were identified in the L. monocytogenes strains, which form the basis for the high pathogenicity of L. monocytogenes. Although the strains analyzed are from different environments and have evolved unique genes to cope with various environmental pressures, they all isolated from beef, and thus share many common features in their evolutionary process. These common features allowed us to identify potential target genes, which were further explored to discover novel targets. Specificity tests and artificial contamination experiments confirmed that the bglF_2 gene holds promise as a specific molecular target for detecting L. monocytogenes strains in beef samples. This study further enhances our understanding of the pathogenicity and adaptability of L. monocytogenes, while providing significant reference value for the development of specific molecular detection targets for this pathogen.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Adnan M.Siddiqui A. J.Noumi E.Hannachi S.Ashraf S. A.Awadelkareem A. M.. (2022). Integrating network pharmacology approaches to decipher the multi-target pharmacological mechanism of microbial biosurfactants as novel green antimicrobials against Listeriosis. Antibiotics (Basel) 12:5. doi: 10.3390/antibiotics 12010005, PMID: 36671206 PMC 9854906 · doi ↗ · pubmed ↗
- 2Asghar A.Khalid A.Baqar Z.Hussain N.Saleem M. Z.Sairash. (2024). An insights into emerging trends to control the threats of antimicrobial resistance (AMR): an address to public health risks. Arch. Microbiol. 206:72. doi: 10.1007/s 00203-023-03800-9, PMID: 38252323 · doi ↗ · pubmed ↗
- 3Ata N. S.Abuelnaga A. S. M.Atta N. S.Hedia R. M.Elgabry E. A.Ibrahim E. S.. (2017). Existence and virulence designation of Listeria Monocytogenes in retail chilled pork byproducts in Cairo porcine markets with trials of using Lactobacillus probiotic as anti-Listerial meat Perservative. IOSR J. Environ. Sci. Toxicol. Food Technol. 11, 19–23. doi: 10.9790/2402-1105041923 · doi ↗
- 4Buszewski B.Rogowska A.Pomastowski P.Złoch M.Railean-Plugaru V. (2017). Identification of microorganisms by modern analytical techniques. J. AOAC Int. 100, 1607–1623. doi: 10.5740/jaoacint.17-0207, PMID: 28703095 · doi ↗ · pubmed ↗
- 5Chowdhury B.Anand S. (2023). Environmental persistence of Listeria monocytogenes and its implications in dairy processing plants. Compr. Rev. Food Sci. Food Saf. 22, 4573–4599. doi: 10.1111/1541-4337.13234, PMID: 37680027 · doi ↗ · pubmed ↗
- 6Colomba C.Rubino R.Anastasia A.Palermo G.Lo P. D.Abbott M.. (2020). Postpartum listeria meningitis. ID Cases 21:e 00896. doi: 10.1016/j.idcr.2020.e 00896, PMID: 32670794 PMC 7347977 · doi ↗ · pubmed ↗
- 7Dapgh A.Salem R. L. (2022). Molecular detection of Listeria monocytogenes in Milk and some Milk products. Int. J. Vet. Sci. 11, 514–519. doi: 10.47278/journal.ijvs/2021.128 · doi ↗
- 8Di H.Ye L.Yan H.Meng H.Yamasak S.Shi L. (2014). Comparative analysis of CRISPR loci in different Listeria monocytogenes lineages. Biochem. Biophys. Res. Commun. 454, 399–403. doi: 10.1016/j.bbrc.2014.10.018, PMID: 25445602 · doi ↗ · pubmed ↗