De novo assembly and annotation of Tetradesmus sp. strain 198, a green algal from dryland soil
Veronica Malavasi, Núria Beltran-Sanz, Elena Catelan-Carphio, Alessandro Grapputo, David J. Eldridge, Samantha Travers, Fernando T. Maestre, Francesco Dal Grande

TL;DR
This paper presents the genome assembly and gene annotation of a green algal strain with potential for carotenoid production.
Contribution
The study provides a high-quality genome assembly and gene annotation for Tetradesmus sp. strain 198.
Findings
The genome assembly has a total length of 149 Mbp with a BUSCO completeness of 91%.
The N50 value of the assembly is 783 kbp, indicating good contiguity.
A total of 19,841 genes were annotated in the genome.
Abstract
Tetradesmus sp. strain 198 is a microalga with potential for the biotechnological production of carotenoids. In this study, we have sequenced the genome, obtaining a total contig-level genome assembly length of 149 Mbp. The BUSCO completeness was 91%, the N50 was 783 kbp, and the total number of annotated genes was 19,841.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
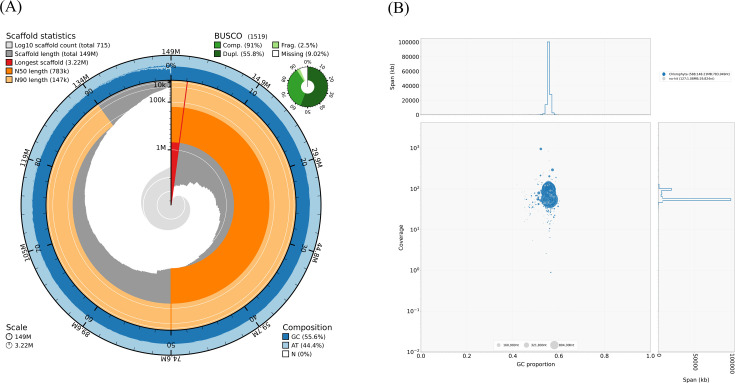 Fig 1
Fig 1- —Università degli Studi di Padovahttp://dx.doi.org/10.13039/501100003500
- —European Research Councilhttp://dx.doi.org/10.13039/501100000781
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAlgal biology and biofuel production · Genomics and Phylogenetic Studies · Microbial Community Ecology and Physiology
ANNOUNCEMENT
Tetradesmus sp. strain 198, a newly described strain of the Chlorophyceae class within the Scenedesmaceae family, has been identified as belonging to the Tetradesmus bajacalifornicus clade based on reference 1. It was isolated from a bare soil sample in an arid region of Australia’s east coast (34.71686, 144.77065), during the Biodesert global dryland survey (2). The strain was cultivated in an Erlenmeyer flask containing 50 mL of modified Bold Basal Medium (3) under continuous light at 22°C and 100 photons m^−2^ s^−1^ for 2 weeks. DNA extraction was performed using the NucleoSpin Plant II kit (Macherey-Nagel, Düren, Germany) according to the manufacturer’s instructions. Two sequencing libraries were prepared: a Nanopore long-read library, sequenced on the ONT MinION system (15 Gbp of raw reads) and an Illumina short-read library, sequenced on the NovaSeqX Plus platform (6.75 Gbp of raw paired-end reads).
Nanopore reads were base-called using Guppy v.7.1.4 with the fast model (400 bps), R10.4.1 MinION Flow Cells (FLO-MIN114), and the SQK-LSK114 library preparation kit. Reads with a quality score above eight were analyzed with Nanoplot v.1.28.1 (4). The summary statistics generated by Nanoplot indicated a total read count of 3,548,659, with a read length N50 of 5,117 bp. Long reads were then assembled into contig using Flye v.2.9 (5).
Illumina short reads were trimmed with Fastp v.0.23.4 (6), applying base correction, low complexity filtering, and maintaining a minimum read length of 100 bp. The report generated by Fastp shows 41 Mbp of total paired-end reads, with a mean length of 149 bp. Prior to and following the trimming process, the quality of the Illumina raw reads was evaluated using FASTQC v.0.11.9 (7). The trimmed Illumina reads were then mapped to the contig-level assembly using BWA-MEM v.0.7.17 (8), and the base-level accuracy was further improved by an iteration of short-read polishing using Pilon v.1.24 (9). We further evaluated potential contamination with Blobtools v.1.1.1 (10) utilizing the generated mapping files and a BLASTN v.2.14.0 (11) against the NCBI nt database (https://www.ncbi.nlm.nih.gov/). Contamination and organelles were then extracted from the main assembly.
The summary of the main characteristics of the genome assembly is shown in the Snailplot performed with Blobtoolkit v.4.2.1 (12, see Fig. 1). The final assembly consists of 715 contigs, with a total length of 149 Mbp, an N50 of 783 kbp, a guanine-cytosine (GC) content of 55.56%, and a BUSCO completeness of 91%. Coverage was evaluated with QualiMap v.2.3 (13), resulting in 30× and 89× for long and short reads, respectively.
(A) SnailPlot and (B) BlobPlot analysis of Tetradesmus sp. strain 198 genome.
Masking of the genome with RepeatModeler v.2.0.5 (14) and RepeatMasker v.4.1.0 (15) showed a repeat content of 19%. Gene annotation identified 19,841 genes using GeMoMa v.9 (16), based on seven Chlorophyta reference genomes from NCBI RefSeq.
Default parameters were used except where otherwise noted.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Mai XC, Shen CR, Liu CL, Trinh DM, Nguyen ML. 2023. “DNA signaturing” database construction for Tetradesmus species identification and phylogenetic relationships of Scenedesmus-like green microalgae (Scenedesmaceae, Chlorophyta). J Phycol 59:775–784. doi:10.1111/jpy.1335437261838 · doi ↗ · pubmed ↗
- 2Maestre FT, Eldridge DJ, Gross N, Le Bagousse-Pinguet Y, Saiz H, Gozalo B, Ochoa V, Gaitán JJ. 2022. The BIODESERT survey: assessing the impacts of grazing on the structure and functioning of global drylands. Web Ecol 22:75–96. doi:10.5194/we-22-75-2022 · doi ↗
- 3Bischoff HW, Bold HC. 1963. Phycological studies IV. Some soil algae from enchanted rock and related algal species, p 95. University of Texas Publication no.6318, Austin, Texas, USA.
- 4De Coster W, Rademakers R. 2023. Nano Pack 2: population-scale evaluation of long-read sequencing data. Bioinformatics 39:btad 311. doi:10.1093/bioinformatics/btad 31137171891 PMC 10196664 · doi ↗ · pubmed ↗
- 5Kolmogorov M, Yuan J, Lin Y, Pevzner PA. 2019. Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol 37:540–546. doi:10.1038/s 41587-019-0072-830936562 · doi ↗ · pubmed ↗
- 6Chen S, Zhou Y, Chen Y, Gu J. 2018. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34:i 884–i 890. doi:10.1093/bioinformatics/bty 56030423086 PMC 6129281 · doi ↗ · pubmed ↗
- 7Wingett SW, Andrews S. 2018. Fast Q screen: a tool for multi-genome mapping and quality control. F 1000 Res 7:1338. doi:10.12688/f 1000 research.15931.230254741 PMC 6124377 · doi ↗ · pubmed ↗
- 8Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. ar Xiv 3:13033997. doi:10.48550/ar Xiv.1303.3997 · doi ↗