Report on Witteveen-Kolk syndrome caused by large fragment deletion in the 15q24.1 - q24.2 region in infants with early onset and literature review
Hanlin Tang, Jiaxuan Xu, Ling Ge, Qunting Gao, Xinlu Tan, Qicheng Qiao, Ruihan Liu, Qingxia Kong, Qiubo Li, Xiufang Jiang

TL;DR
This paper reports a rare case of Witteveen-Kolk syndrome in a 4-month-old infant caused by a large chromosomal deletion, emphasizing the importance of early diagnosis and intervention.
Contribution
The first reported case of WITKOS caused by a large fragment deletion in the 15q24.1 - q24.2 region.
Findings
The infant showed early onset symptoms including facial abnormalities and developmental delays.
Early rehabilitation improved the infant's motor and language abilities significantly.
The case highlights the need for clinical awareness and management of WITKOS.
Abstract
WITKOS is a rare neurodevelopmental disorder caused by heterozygous loss-of-function variants in the 15q24.1 - q24.2 region, includeing switch-insensitive 3 transcription regulator family member A (SIN3 A). Its incidence rate is extremely low. According to the current limited global data statistics, only a few cases occur per million population. Most cases concentrate in childhood and adolescence. During this stage, the rapid physiological changes of the body seem to be closely related to the triggering of the disease. Its characteristics include unique facial features, intellectual and motor developmental delay, and short stature. This paper reports a case of WITKOS in a 4-month-old infant caused by large-fragment copy number variation in the 15q24.1 - q24.2 region of the chromosome that encompasses the SIN3 A gene. Gene mutations lead to abnormal functions of key proteins, which…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
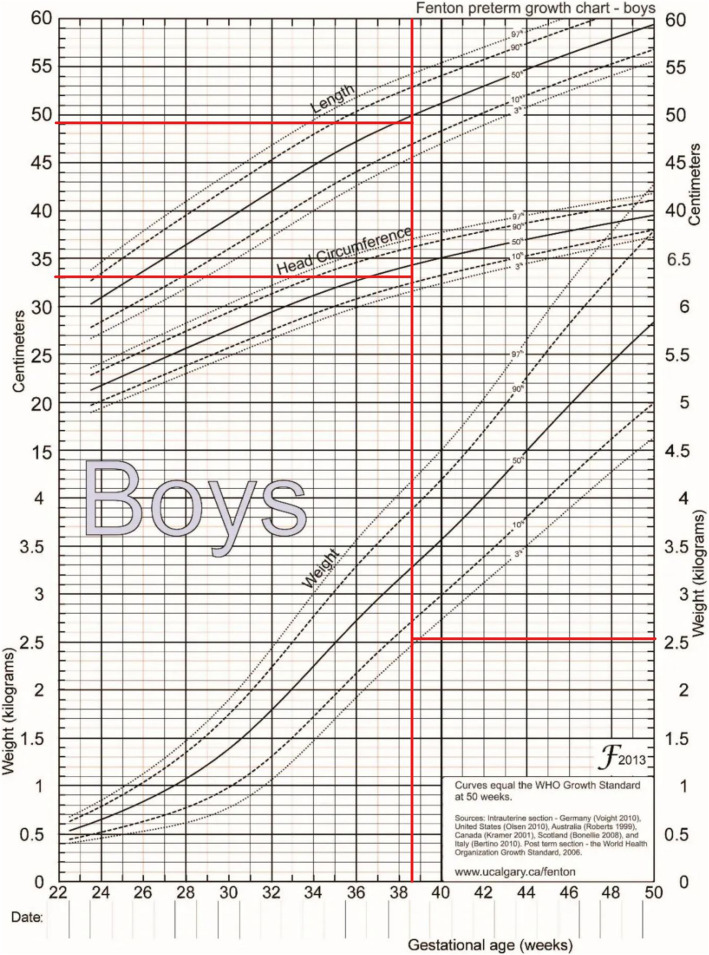 Figure 1
Figure 1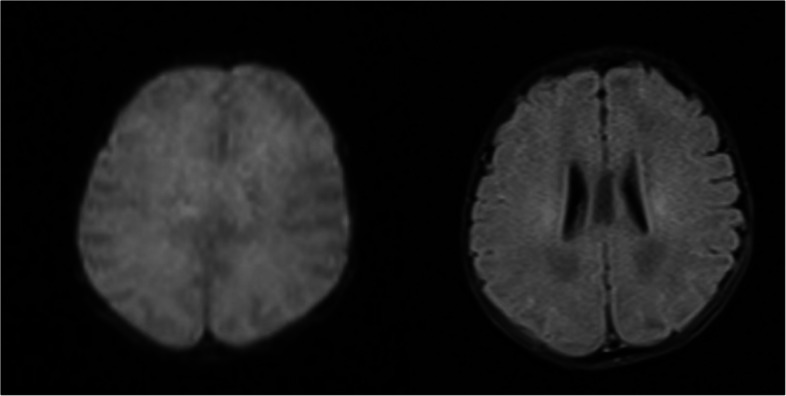 Figure 2
Figure 2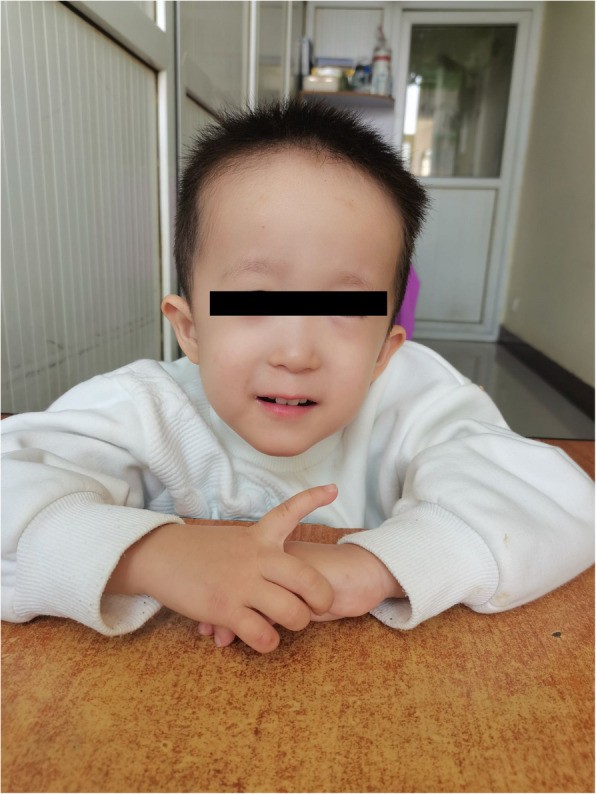 Figure 3
Figure 3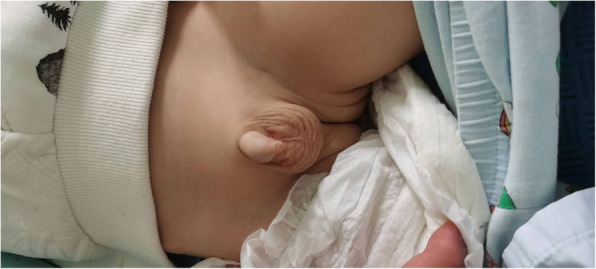 Figure 4
Figure 4 Figure 5
Figure 5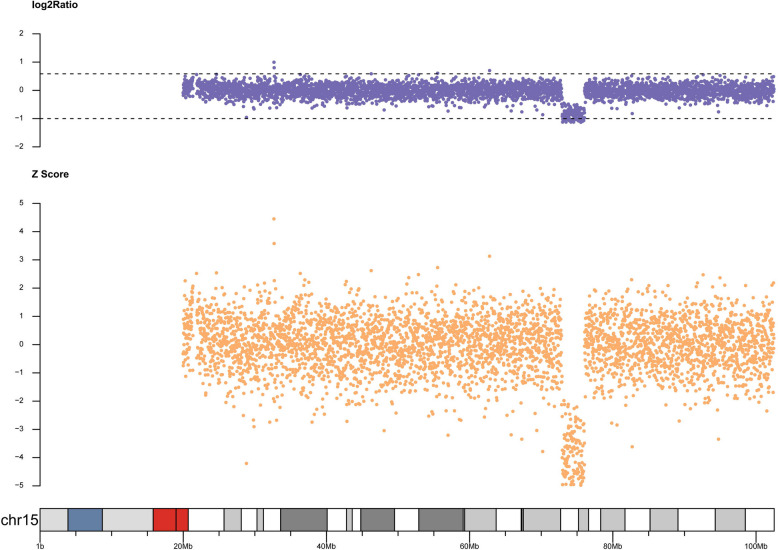 Figure 6
Figure 6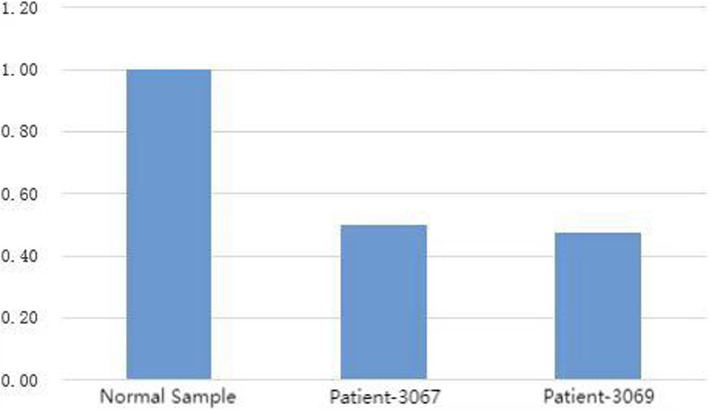 Figure 7
Figure 7- —http://dx.doi.org/10.13039/100016697Youth Innovation Technology Project of Higher School in Shandong Province
- —http://dx.doi.org/10.13039/501100013064Key Research and Development Program of Jiangxi Province
- —http://dx.doi.org/10.13039/501100013064Key Research and Development Program of Jiangxi Province
- —http://dx.doi.org/10.13039/501100013064Key Research and Development Program of Jiangxi Province
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomic variations and chromosomal abnormalities · Genetics and Neurodevelopmental Disorders · Congenital Ear and Nasal Anomalies
Background
WITKOS is a rare neurodevelopmental disorder that was first described in 2016, with distinctive facial features (such as a broad and high forehead, small mouth, thin upper lip, pointed chin, and downward-slanting palpebral fissures), microcephaly, short stature, mild intellectual disability (ID), cognitive and motor developmental delays, and corpus callosum dysplasia and ventricular enlargement on brain Magnetic Resonance Imaging (MRI), etc. [1].The disease is caused by SIN3 A heterozygous loss-of-function variations and is also characterized by various intellectual disabilities and developmental delays. WITKOS is inherited in an autosomal dominant manner, and most patients are sporadic. In terms of the existing literature, up to now, the reported research has mainly focused on the patient groups of older children, adolescents, and adults. There has been no relevant report on the case of WITKOS caused by large - fragment deletion in infancy. This report aims to fill this gap and expand the case data of WITKOS. This study reports a case of WITKOS caused by copy number variation of SIN3 A in a 4-month-old infant, summarizes the characteristics of its gene phenotype, and provides some diagnostic and therapeutic ideas for clinicians in screening for developmentally deviated young children. It is hoped that through this study, more effective treatment options for WITKOS can be provided to improve the prognosis of patients.
Case presentation
The patient was a 4-month and 2-day-old male infant who presented with"unstable neck holding"on November 12, 2021. Physical examination showed a weight of 6.0 kg (−1 to −2SD), a height of 60.1 cm (−2SD), and a head circumference of 41.7 cm (0SD). The special facial features included a wide and high forehead, a low nasal bridge, full and downward-slanting eyelids, a small mouth, a small mandible, and low-set ears. There were no abnormalities in the heart and lungs. Genital examination revealed a short penis and left cryptorchidism. Motor examination showed that the muscle tone of the limbs was slightly low during the first physical examination, but on December 17, 2021, there was a decrease in muscle tone, a positive scarf sign, poor head control, head retroflexion when pulled up from the supine position, and unstable neck holding, but the ability to lift the head was present. Both hands could be centered and could suck the hands. Cognitive and reactive language examination showed few facial expressions, little interaction, poor repetitive visual tracking, rarely crying, few babbling sounds, inability to be amused to laugh, and no awareness of grasping objects. The child was the second pregnancy and second birth (G2P2). The mother underwent cesarean section at 38 + 5 weeks of gestation due to"polyhydramnios, pregnancy with uterine scar, vaginitis, and mild anemia"during pregnancy. At birth, the weight was 2.53 kg (3.8%), the height was 49.0 cm (33.7%), the head circumference was 33.0 cm (17.5%), and the Apgar score was 10 at 1, 5, and 10 min, respectively, indicating a small-for-gestational-age infant (see the FENTON curve chart, Fig. 1). After birth, the child was fed with a mixed diet and had difficulty in feeding, with each breastfeeding session lasting more than half an hour, and the growth rate was slow. On the first day after birth, the child was hospitalized in the NICU due to"neonatal amniotic fluid swallowing syndrome and neonatal ABO blood group incompatibility hemolytic disease", and the jaundice reached a maximum of 17.0 + mg/dl. On the third day of hospitalization, brain MRI plain scan and diffusion-weighted imaging showed no obvious abnormalities (Fig. 2), and the child was discharged after improvement three days later. On July 27, 2021, the thyroid function test was normal. On November 11, 2021, during the 4-month physical examination, the developmental test score was 62, the bone density was − 0.8, and the neuropsychological developmental test for children aged 0–6 years showed a developmental quotient of 62 (the normal value is between 85 and 115), with a gross motor score of 37, fine motor score of 74, adaptive ability score of 50, language score of 87, and social behavior score of 62. After diagnosis, the child received early rehabilitation intervention locally. On October 6, 2022, during a telephone follow-up at 15 months of age, the child's height was about 80 cm and the weight was about 10 kg. The progress of gross motor skills was faster than that in other functional areas. The child could not walk independently but could stand with support for a short time, could walk and bend over to pick up objects while supported, could get on and off the bed independently, and could play with toys independently. The muscle tone was low, the child could hold the bottle to drink milk, and the femoral angle was 180°. The progress of fine motor skills, cognition, and language was slower, there were unconscious slapping movements, the child could not understand one-step instructions, but could call mom and dad. The father's blood type was A, Rh unknown, and he was healthy. The mother's blood type was O, Rh positive, and both parents had medium stature. They were not closely related, and had an older sister who was 3 years and 8 months old. The sister was hospitalized in the neonatal intensive care unit of the local hospital for 10 days after birth and is currently healthy and developing normally, with no similar family history.In the follow-up on October 12, 2024, the child can currently walk and run independently and can say short sentences but not long ones, After rehabilitation intervention, the overall development score of the child has increased from the initial 217 points to 278 points (sensory perception 15 - 22, gross motor skills 79 - 91, fine motor skills 22 - 28, self - care ability in daily life 42 - 50, language communication 36 - 45, cognition 11 - 19, sociality 12 - 23), and the overall improvement effect is good. Figure 3 shows the photo of the child during the follow-up. It can be seen that the child still has a wide and high forehead, a low nose bridge, full and drooping eyelids, small lips, a small mandible, and a low ear position. The cryptorchidism of the child has been effectively improved. At present, no obvious abnormal manifestations have been observed. See Fig. 4.Fig. 1. Schematic diagram of the FENTON curve chart of the patient, and the intersection point represents the patient's situationFig. 2Brain MRI plain scan and diffusion-weighted imaging of the patientFig. 3The facial features of this child: a wide and high forehead, a low nose bridge, full and drooping eyelids, small lips, a small mandible, and a low ear positionFig. 4It can be seen that the cryptorchidism of the child has been effectively improved
Chromosome examination
After obtaining informed consent from the parents of the patient, blood samples were collected for chromosome aneuploidy and microdeletion or microduplication examination. The report date was December 09, 2021, and the examination results were as follows:
Chromosome aneuploidy: The composition of the examined chromosomes was 46, XY, and the number of chromosomes was normal. See Fig. 5. Chromosome microdeletion or microduplication: CNV detection revealed pathogenic CNV variations. Through copy number variation verification (qPCR - SYBR Green I dye method), a deletion variation was found in one region. Interpretation of the abnormal result: Abnormal type: deletion (loss).Fig. 5. Genome-wide detection result diagram of the examined patient: The composition of the examined chromosomes is 46, XY, and the number of chromosomes is normal, This information provides a basis for subsequent analysis and rules out the possibility that the disease is caused by abnormal chromosome numbers
Pathogenicity: Pathogenic. Chromosome location: 15q24.1 - q24.2. Start - end position of the abnormal fragment (UCSC hg19): 72,929,607 - 76,069,619. Size of the abnormal fragment (Mb): 3.140. The copy number distribution of the examined person on chr15 is shown in Fig. 6.Fig. 6. Copy number distribution map of the examined patient on chr15 chromosome. Chromosome Location: 15q24.1-q24.2 Start-End Position of the Abnormal Fragment (UCSC hg19): 72,929,607 - 76,069,619 Size of the Abnormal Fragment (Mb): 3.140, This change in copy number is closely related to the patient's disease state and may be an important genetic factor leading to the symptoms of WITKOS
Using fluorescence quantitative PCR to analyze the data further, we selected suspicious fragments similar to the proband's phenotype and then did Q - PCR experiments to verify the region, excluding false positives of second-generation sequencing and ensuring result accuracy. The specific experimental process is as follows: The experimental instrument used was the 7300Plus Real - Time PCR System, and the reagent used was UltraSYBR Mixture (High ROX). For the PCR system, the reaction mixture included 12.5 μl of enzyme, 10.5 μl of water, 1 μl of genome (with a concentration of 50 ng/μl), and 1 μl of primer (with a concentration of 5 μmol/L). The temperature settings were as follows: an initial step at 95 °C for 10 min, then 38 cycles with each cycle including steps at 95 °C for 15 s, 63 °C for 45 s (with fluorescence detection), then again at 95 °C for 15 s, and 63 °C for 1 min (with fluorescence detection during heating), and after the cycles, additional steps at 95 °C for 15 s and 63 °C for 15 s. The analysis software used was Real - Time PCR Software V2.4, and the final result was obtained. The QPCR verification result is shown in Fig. 7. The main purpose of primer design is to specifically recognize the target DNA sequence and guide DNA polymerase to carry out amplification reactions under appropriate conditions. Primers are short single-stranded DNA sequences that can pair complementarily with specific regions on the template DNA, thereby determining the starting position of amplification.Fig. 7QPCR verification result. Note: The chromosome ranges verified by primers 3067 and 3069 are both partial segments of the detected result chr15: 72,929,607 - 76,069,619. Primer 3067 sequence: Forward: TGTTAGGGGCTTCTCGGGAT; Reverse: TCAGGGGGAGGATTAGGGTC. Primer 3069 sequence: Forward: GCAGTCCCCCTAAGTCCAAG; Reverse: AGAGAATCTGGTGCAGTCGC
The whole exome sequencing analysis of the child showed that no pathogenic or suspected pathogenic variants that could explain the phenotype of this child were found.
The result of the Q - PCR experiment on the detection result was that the copy number was approximately 0.5, and the copy number of a normal sample (diploid) was 1. The experimental result indicated that this patient indeed had a copy number abnormality at 15q24.1 - q24.2. Based on the clinical characteristics of this patient and the examination results, WITKOS was finally diagnosed. Moreover, there is a likelihood that this case may have another coexisting genetic disease, which might also be one of the reasons for the phenotype of the child reported in this paper.
Discussion and conclusions
The genetic analysis of the infant in this study revealed that within the 15q24.1 - q24.2 chromosomal region, several genes were identified, with SIN3 A being the key pathogenic gene. Other genes such as CYP11 A1, STRA6, PML, HCN4, LOXL1, and MPI were also present, although their direct relevance remains to be further explored. The related pathogenic gene with the same dominant inheritance pattern as SIN3 A is HCN4. Mutations in the HCN4 gene can manifest as cardiovascular symptoms and language developmental delay. The child has not been detected with reduced cardiovascular system function but shows language developmental delay, which may contribute to the phenotype of this child. According to the Clingen report, SIN3 A's haploid dose score is 3. Enough evidence shows dose sensitivity is related to the phenotype. So, the deletion in the 15q24.1 - q24.2 region including SIN3 A may have caused the child's phenotype.
We searched but didn't find prior reports of WITKOS due to large - fragment deletions like ours. This offers a new perspective on how such gene changes impact WITKOS symptoms. This child in our report did not show seizures, abnormal vision and hearing, abnormal brain MRI, or behavioral problems. (Table 1) However, most children with WITKOS caused by other single-gene abnormalities have these abnormalities. We wonder if the loss of large gene fragments has led to the non-appearance of these abnormalities. In the follow-up, we need to pay close attention. At the same time, more cases of large gene fragment deletions are needed to prove whether our conjecture is correct. Table 1. Comparison of the characteristics of this patient with reported patientDemographic DataGenetic DataMedical CharacteristicsMaleFemaleAgeChromosomeMutationSpecific Facial Features(broad forehead, small mouth, etc.)Small—for—Gestational—Age InfantIntellectual Disability or Developmental DelayFeeding DifficultiesDecreased Muscle ToneThis study (n = 1)1-4 months and 2 daysChr15: 72,929,607–76069619NR11111Narumi-Kishimoto Y (n = 1) [2]-17 yearsNRc.848dup111NR-Balasubramanian M et al. (n = 28) [3]1711Average 8.2 yearsNR1.c.1462del,p.(Val488Leufs7)2.c.2764C > T,p.(Arg922*)3.c.588del,p.(Asn197Metfs4)4.c.377C > T,p.(Ala126Val)5.c.1245_1246del,p.(Asn415Lysfs24)6.c.775dup,p.(His259Profs47)7.c.824del,p.(Pro275Hisfs12)8.c.2248_2251del,p.(Leu750Metfs43)9.c.2339del,p.(Ala780Glyfs14)10.c.889C > T,p.(Gln297*)11.c.1715_1722delinsCCCAAGTGTA,p.(Gly572Alafs11)12.c.3323C > G,p.(Ser1108)13.c.3490A > T,p.(Lys1164*)14.c.46C > T,p.(Gln16*)15.c.3310C > T,p.(Arg1104*)16.c.2809_2810del,p.(Lys937Glufs2)17.c.1489_1499del,p.(Arg497Cysfs13)18.c.3317 T > C,p.(Met1106Thr)19.c.1675C > T,p.(Arg559*)20.c.3303C > G,p.(Tyr1101*)21.c.1570_1577del,p.(Tyr524Valfs26)22.c.2185_2186del,p.(Leu729Glyfs8)23.c.3441_3445del,p.(Lys1148Argfs12)24.c.1318_1737dup;p.(Val580Lysfs35)25.c.1773G > A,p.(Trp591*)26.c.463A > G,p.(Lys155Glu)27.c.2803C > T,p.(Arg935*)28.c.1888dup,p.(Ile630Asnfs17)28NR131512WitteveenJS et al. (n = 15) [1]77(1 person unknown)4–45 yearsChr151. Del 75.60–76.102. Del 75.60–76.103. Del 75.60–76.024. Del 75.60–75.955. c.803dup; p.Leu269fs6. c.1010_1013del; p.Lys337Serfs7–9. c.1759_1759delT; p.Ser587fs10. c.2955_2956del; p.Glu985fs11–13. c.3310C > T; p.Arg110414. Del 75.53–75.8015. Del 75.59–76.0915NR12NR2Adam Jacobson, MD et al. (n = 2) [4]-27 years2 yearsNRc.1657C > T, p.R5532NR2NRNRLinde C. M. van Dongen et al. (n = 5) [5]3214 years10 years12 years23 years10 yearsChr15(GRCh37)1.c.1010_1013del,2.c.2955_2956del,3.c.803dup,4.c.1675C > T,5.c.1488_1498delNRNR5NRNRTotal (n = 52)2823472341615Medical characteristicsSeizuresBehavioral ProblemsShort StatureMicrocephalyVision AbnormalitiesHearing AbnormalitiesBrain MRI AbnormalitiesThis study (n = 1)--1----Narumi-Kishimoto Y (n = 1) [2]-1--NR-NRBalasubramanian M et al. (n = 28) [3]41264464WitteveenJS et al. (* n* = 15) [1]3676NR47Adam Jacobson, MD et al. (n = 2) [4]NRNRNRNR2NRNRLinde C. M. van Dongen et al. (n = 5) [5]35NRNR243Total (n = 52)1024141081414NR Not Reported^*^In this case, the infant did not present with seizures, abnormal vision and hearing, abnormal brain MRI findings, or behavioral problems
In mammals, the eukaryotic chromatin structure is made up of histone octamers of the core histones H2 A, H2B, H3, and H4, which form nucleosomes to ensure that the DNA is compressed. Post-translational modification of the N-terminus of histones, especially acetylation, plays a decisive role in the dynamic regulation of gene transcription. histone deacetylation is performed by histone deacetylase (HDAC) HDAC1 and HDAC2 and is dependent on complexes containing Sin3, MecP2, NuRD, and CoREST. Recruitment of the Sin3 corepressor module leads to deacetylation of histones H3 and H4. SIN3 A encodes a transcriptional regulatory protein associated with scaffold formation in core histone deacetylase complexes [6]. The formed SIN3-HDAC complex serves as a cofactor scaffold and promotes the histone H3 and H4 deacetylation process by recruiting Sin3 corepressor modules [6–8]. liu and pile et al. [9] believe that SIN3 is an important epigenetic regulator and is directly linked to methionine metabolism and histone modification. SIN3 A plays an integral role in the development of the nervous system. SIN3 A is required for cortical progenitor cell differentiation, and decreased SIN3 A expression will lead to altered cortical neuron identity. In addition, SIN3 A expression also affects cortical differentiation and axon elongation. Downregulation of SIN3 A expression increases the length and number of axons that cross the midline along the corpus callosum pathway, while decreased expression results in axon elongation deviating from the original route. Functional knockout of SIN3 A in vivo resulted in reduced neurogenesis, altered neuronal identity, and abnormal cortical projection in developing mice. In mammals, there are two genes responsible for encoding the main factors of the Sin3 complex, namely SIN3 A and SIN3B [10]. If SIN3 A is absent or has other abnormalities, WITKOS with special facial features and clinical symptoms such as intellectual impairment may occur.
The SIN3 A gene is in the 15q24 segment and is a key part of the transcriptional corepressor complex. It was once seen as important for the 15q24 microdeletion syndrome [11]. The Sin3 A transcription protein, with a molecular scaffold function, offers a platform for functional transcription factors. It has six domains, including a histone interaction domain (HID) that can recruit core complexes like HDAC, histone-binding protein RBAP46/48, stabilizer SAP18/20, and SDS3 [12]. Sin3 A is crucial for transcription regulatory factors like HDAC [1]. It's one of the main genes encoding the Sin3 complex, and its encoded protein is related to various physiological processes. In nervous system development, SIN3 A is essential. WITK0S results from SIN3 A function loss, and its haploinsufficiency causes WITKOS [1, 3]. Regarding SIN3 A's effect on patient methylation status, studies have different conclusions. Suzuki et al. [13] and Liu & Pile [9] think its haploinsufficiency may affect WITKOS patients'methylation. Kawai [14] found no methylation difference. Narumi-Kishimoto Y [2] detected no abnormal DNA methylation, believing SIN3 A frameshift mutations don't impact it. Jet Coenen - van der Spek [15] established a WITKOS DNA methylation epigenetic probe, diagnosed an undiagnosed case, and suggested SIN3 A haploinsufficiency may not be the only factor.
Witteveen et al. [1] reported that some WITKOS patients had motor disorders (gross and fine motor skills), which is a developmental coordination disorder (DCD) diagnosed by motor coordination difficulties. DCD also exists in other neurodevelopmental disorder children, so it's often not recognized and diagnosed by medical professionals [16]. The patient in this report, like Narumi-Kishimoto Y [2]'s case, was a small-for-gestational-age infant without microcephaly, vision/hearing abnormalities, brain MRI abnormalities, or seizures. Table 1 [1–5] summarized reported WITKOS patients'characteristics. This child was diagnosed after 4 months with intellectual developmental delay (poor cognitive understanding, no active language), motor developmental delay (decreased muscle tone, delays in gross and fine motor skills), short stature, and special facial features. After early rehabilitation training, the child's motor ability improved. Clinically, similar patients need early assessment and rehabilitation. For those with early feeding difficulties, provide feeding guidance and observe growth. Identify and manage behavioral problems early and follow up for ASD. The disease has a poor prognosis and requires long-term intervention. Despite its low incidence and sporadic nature, due to its autosomal dominant inheritance, parents of an affected child should be examined. If a parent has a 15q24 microdeletion or SIN3 A gene abnormality, the offspring has a 50% inheritance chance. Third-generation in vitro fertilization technology (PGD) is recommended for intervention. Future research should focus on SIN3 A function, treatment methods, and improving patient management and awareness. Comparing with 15q24 microdeletion syndrome children, this WITKOS child has similar growth, intellectual, facial, and muscle tone characteristics, making clinical distinction difficult and highlighting the need for gene testing.
Our WITKOS patient and children with 15q24 microdeletion syndrome have some similar characteristics. Magoulas PL [17] found that SIN3 A gene problems can cause special symptoms, which means SIN3 A is important. Both diseases have growth retardation, abnormal intellectual development, special facial features, and abnormal muscle tone. These similarities make it hard to tell them apart. In clinical work, we need to consider many things and do more gene testing to know if a child has WITKOS.
Future research should focus on understanding SIN3 A better. We need to know how it works in the nervous system and how it interacts with other genes. Also, we should develop better treatments for WITKOS, like targeted drugs or gene therapy. At the same time, we need to improve how we monitor and manage WITKOS patients and make doctors and people know more about this disease to help patients live better.
In conclusion, This case is the first report of WITKOS caused by a large - fragment deletion in infancy, which expands the understanding of the genetic mechanism and early manifestations of the disease. It fills the gap in the onset of infancy, suggests the characteristics of early symptoms, helps in earlier identification and intervention, and changes the disease process. Meanwhile, it enriches the gene - phenotype association study, facilitates the precise location of the pathogenic region of the SIN3 A gene, and provides a theoretical basis for treatment. Our research indicates that for this particular case, clinical observation and genetic testing were crucial for diagnosis. Through these means, the patient's condition was identified, and subsequent rehabilitation training was implemented. The patient seemingly benefited from these interventions, with potential improvements in prognosis and quality of life observed. Nevertheless, it is essential to recognize that these conclusions are drawn from a single case and cannot be generalized. Future research with larger sample sizes is needed to validate these initial findings.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Adam Jacobson, MD, Brenda L. Bohnsack, MD, PHD. Anterior megalophthalmos in sisters with Witteveen-Kolk syndrome [J]. Journal of AAPOS,2022,26(3):148–150.10.1016/j.jaapos.2022.01.00335144002 · doi ↗ · pubmed ↗
- 2van Dongen 1,Ellen Wingbermühle,Alexander J.M.Dingemans,et al.Behavior and cognitive functioning in Witteveen-Kolk syndrome[J].Medical Genetics,2020;182A:2384–2390.10.1002/ajmg.a.61775 PMC 754040932783353 · doi ↗ · pubmed ↗
- 3Latypova X,Vincent M,Molle A,et al. Haploinsufficiency of the Sin 3/HDAC corepressor complex member SIN 3B causes a syndromic intellectual disability/autism spectrum disorder[J]. Am J Hum Genet. 2021;108(5):929–941.10.1016/j.ajhg.2021.03.017PMC 820616633811806 · doi ↗ · pubmed ↗
- 4Coenen-van der Spek J, Relator R, Kerkhof J, et al. DNA methylation episignature for Witteveen-Kolk syndrome due to SIN 3A haploinsufficiency. Genet Med. 2023;25(1):63–75. 10.1016/j.gim.2022.10.004.10.1016/j.gim.2022.10.00436399132 · doi ↗ · pubmed ↗
- 5Harris SR, Mickelson EC, Zwicker JG. Diagnosis and management of developmental coordination disorder[J]. CMAJ Can Med Assoc J. 2015;187(9):659–665.10.1503/cmaj.140994 PMC 446792926009588 · doi ↗ · pubmed ↗
- 6Magoulas PL, El-Hattab AW. Chromosome 15q 24 microdeletion syndrome. Orphanet J Rare Dis. 2012;7:2. Published 2012 Jan 4. 10.1186/1750-1172-7-2.10.1186/1750-1172-7-2PMC 327544522216833 · doi ↗ · pubmed ↗