Faster genetic mapping of complex traits in C. elegans
Stefan Zdraljevic, Laura Walter-McNeill, Alex Lee, Joshua Bloom, Leonid Kruglyak

TL;DR
Researchers found that RNAi can replace a specific allele to speed up genetic mapping in C. elegans.
Contribution
RNAi knockdown is shown as a viable alternative to a specific allele for generating recombinant populations in C. elegans.
Findings
Genome-wide allele frequencies were similar between populations using fog-2(q71) and RNAi.
RNAi can be used to rapidly generate recombinant populations for genetic mapping.
This method allows more flexibility in experimental design for genetic studies.
Abstract
Caenorhabditis elegans is a tractable model system that enables the identification of genetic determinants that underlie phenotypic variation. Over the years, new approaches have been developed to lower the cost of and expedite genetic mapping in this model system. The ce X-QTL approach uses the fog-2 ( q71 ) allele to create obligate outcrossing recombinant populations for selection and sequencing experiments. Here, we tested whether the fog-2 ( q71 ) allele is essential to the ce X-QTL approach by comparing crosses between the N2 and XZ1516 strains using either fog-2 ( q71 ) or fog-2 RNAi knockdown to facilitate outcrossing. The genome-wide allele frequencies of the bulk recombinant populations derived from these two methods were largely similar. These results demonstrate that fog-2 RNAi is a viable alternative for rapidly generating recombinant populations, allowing…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
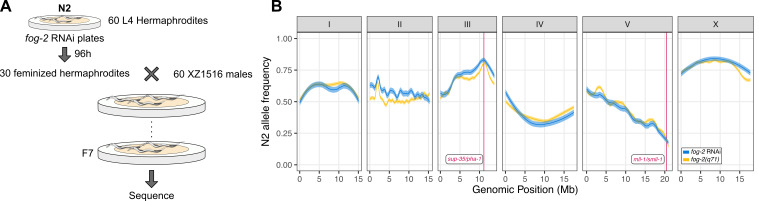 Figure 1
Figure 1|
Strain |
Allele name |
Genotype |
Description |
|
|
Wild
| ||
|
|
Wild
| ||
|
|
|
|
|
|
|
|
|
|
|
Strain |
Allele name |
Genotype |
Description |
|
|
|
|
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic and phenotypic traits in livestock · Genetic Mapping and Diversity in Plants and Animals
Description
Caenorhabditis elegans * has proven to be an exemplary model system to probe how genetic factors shape traits (Andersen & Rockman, 2022). The success of * C. elegans * as a model system for quantitative genetics can be attributed to its short generation time, relative ease of setting up crosses, well-annotated high-quality genome sequence, and a wide variety of molecular and population genetics tools (Brenner, 1974; C. elegans Sequencing Consortium, 1998; Cook et al., 2017).
A typical experiment to identify genetic factors underlying trait variation in the population involves constructing a panel of recombinant inbred lines derived from two phenotypically different parental strains (Andersen et al., 2015; Rockman & Kruglyak, 2009). However, constructing, genotyping, and phenotyping these panels is laborious and costly. We recently developed an alternative approach, ce X-QTL, that relies on incorporating the * fog-2 ( q71 ) * loss of function allele into parental strains to make them obligate outcrossers (Burga et al., 2019; Schedl & Kimble, 1988). This allele enables construction of large pools of recombinant progeny that can be subjected to selection regimes to find the genetic basis of phenotypic variation in the population. To further expedite the mapping process, we asked whether the * fog-2 ( q71 ) * allele is required for ce X-QTL.
To answer this question, we introduced the * fog-2 ( q71 ) * allele into two genetically divergent * C. elegans * strains, XZ1516 and N2 . These two strains are incompatible at the * sup-35 / pha-1 * and mll-1/smll-1 toxin-antidote elements (TAs) (Ben-David et al., 2017; Zdraljevic et al., 2024), which served as genomic loci with strong and stereotypical effects on allele frequencies in recombinant populations. Our goal was to compare allele frequencies in a cross between XZ1516 ( * fog-2 ( q71 ) * ) and N2 ( * fog-2 ( q71 ) * ) to those from a cross between N2 and XZ1516 propagated on RNAi plates that knock down the expression of * fog-2 * ( Figure 1A ) (Kamath et al., 2001; Timmons & Fire, 1998). Our rationale for this experiment was that temporary feminization of the parental lines may be sufficient to generate pools of recombinants that could be used for bulk selection experiments.
We sequenced the two resulting recombinant populations after seven generations of intercrossing on * fog-2 * RNAi plates and calculated the N2 and XZ1516 allele frequencies across the genome. We observed the expected depletion of the N2 genotype at the mll-1/smll-1 TA locus on chromosome V in both crosses ( Figure 1B ). Importantly, there wasn't a significant difference in allele frequencies at this locus between the two crosses ( p value= 0.89 at V:20469619). Similarly, we observed a depletion of the non-carrier XZ1516 genotype at the * sup-35 / pha-1 * locus in both crosses, with only a minimal difference between the allele frequencies ( *p * value = 0.03 at III:11119227). The overall patterns of allele frequencies across the genome were very similar in both crosses, with both approaches detecting the same allele frequency distortions on chromosomes I, IV, and X, outside the known TAs.
Our results show that the * fog-2 ( q71 ) * allele is not required for construction of bulk recombinant populations for ce X-QTL experiments. Instead, large recombinant populations can be quickly generated on * fog-2 * RNAi to assess the genetic basis of trait variation. Once constructed, these populations can be taken off * fog-2 * RNAi and maintained as hermaphrodite bulk populations, or, if desired, singled and amplified to construct recombinant inbred line panels. There are, however, limitations to using * fog-2 * RNAi to construct bulk recombinant populations. It is known that there is substantial variation in RNAi sensitivity across the * C. elegans * population, and one would have to assess RNAi sensitivity of the parental lines prior to moving forward with crosses on * fog-2 * RNAi plates (Chou et al., 2024; Paaby et al., 2015; Pollard & Rockman, 2013). Additionally, because RNAi is not fully penetrant in even the most sensitive * C. elegans * isolates, self progeny are expected to arise during the construction of * fog-2 * RNAi-induced recombinant populations, which may reduce the mapping resolution at any given QTL by lowering the overall number of recombination events in the population. Given the limited number of recombination events per generation in * C. elegans * (Rockman & Kruglyak, 2009), we think that these are reasonable tradeoffs given the ease of construction of recombinant populations via * fog-2 * RNAi.
Methods
Strains
N2 and XZ1516 were acquired from the Caenorhabditis elegans Natural Diversity Resource (Cook et al., 2017). The
- E. coli* strains OP50 and HB101 were acquired from the Caenorhabditis Genetics Center (CGC), which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). We previously described the introduction of the fog-2(q71) allele into XZ1516 to construct the QX2538 strain (Zdraljevic et al., 2024). We constructed QX2544[ *fog-2(q71[P17stop]) * in N2] in the same manner.
Strain maintenance
Prior to growth on RNAi-expressing bacteria, all C. elegans strains were maintained at 20°C on modified nematode growth medium (NGMA), containing 1% agar and 0.7% agarose (Andersen et al., 2014) and seeded with OP50 E. coli . Spontaneous XZ1516 males were identified on 10 cm plates and used to establish male cross cultures.
RNAi bacteria
HT115 bacteria expressing fog-2 RNAi in the L4440 plasmid background were acquired from Horizon Biosciences (Catalog#: RCE1182-202300280) (Rual et al., 2004; Timmons & Fire, 1998). Prior to induction, the fog-2 RNAi bacteria were streaked out to single colonies on LB + 100 µg/ml carbenicillin agar plates. Single colonies were grown overnight in 3 ml liquid LB + 100 µg/ml carbenicillin cultures. The following day, these cultures were diluted 1:1000 in LB + 100 µg/ml carbenicillin and grown for 16 hours. Finally, the expression of fog-2 RNA was induced with 1 mM IPTG for four hours. 100 µl of 1000 µl of these cultures were used to seed 6 cm or 10 cm plates, respectively. RNAi plates were dried for two days at room temperature and then transferred to 4ºC. We only used RNAi plates that were seeded within seven days in all experiments.
RNAi sensitivity
To test the susceptibility of the strains to *fog-2 * RNAi, we transferred ~10 N2 and XZ1516 L4 hermaphrodites to *fog-2 * RNAi plates and allowed them to generate progeny. Once the progeny reach L4, we singled ~30 individuals to agar plates. The following day we counted the number of individuals that laid progeny. For both N2 and XZ1516, approximately 5% of the transferred individuals generated progeny, indicating that these strains are highly susceptible to RNAi.
Cross construction
Multiple 6 cm fog-2 RNAi plates were seeded with five N2 L4s and incubated at 20ºC for 96 hours until adults from the next generation were present. Adults that expressed the fog-2 -specific stacked oocyte phenotype were considered to be feminized (Qin & Hubbard, 2015). Thirty feminized N2 animals were crossed to 60 XZ1516 males on fog-2 RNAi plates overnight and assessed for plugs the following day (Noble et al., 2015). Plugged N2 worms were then transferred to three 10 cm fog-2 RNAi plates and allowed to lay progeny for 96 hours. 96 hours later, the resulting population was washed off these 10 cm plates with M9 media. The M9 containing the cross populations were transferred to a 15 ml conical tube and allowed to settle for 10 minutes. After this time ~75% of the supernatant in the conical tube was transferred to a new conical tube. The transferred media contained young larval F2 worms that were then titered to accurately seed the next generation. Approximately 15,000 young larvae were used to seed the subsequent generation with approximately 1000-1500 animals per 10 cm fog-2 RNAi plate. The process was repeated until the F7 generation, at which point the population was frozen for sequencing. The exact same process was performed for the N2 *fog-2(q71) * and XZ1516 fog-2(q71) to minimize the effects of being grown on different food sources.
Sequencing and analysis
Genomic DNA was prepared using Purelink Genomic DNA Mini Kit (invitrogen cat# K1820-01), libraries were constructed using the Nextera XT library kit (illumina cat# FC-131-1024), and sequenced on the NextSeq2000 platform. Demultiplexing was performed on basespace. FASTQ files were aligned to the WS276 genome assembly using bwa mem with default parameters (Li, 2014). GATK ASEReadCounter (v4.6.0.0) was used to count the number of reference and alternate alleles in each of the cross populations. The VCF from the 20220216 CaeNDR release was used to determine the segregating sites in the cross populations. The xQTLStats R package, which uses the methodology described in Huang et al., was used for calculating allele frequencies, standard errors, and p values associated with allele frequency differences between the crosses (Huang et al., 2020). Allele count tables generated by GATK *ASEReadCounter * were loaded into R using the makeCountTables function from the xQTLStats package. We next removed sites with fewer than 10 reads supporting the genotype calls and any sites that had zero counts for the reference or alternate alleles. We next ran the calcAFD function on the experimental allele count tables with the following parameters: sample.size=1e4, sel.strength=.95, bin.width=9000, eff.length=2000. Finally, we ran calcContrastStats to calculate the p values associated with the allele frequency differences between the RNAi and KO experiments. We submitted the sequencing reads to SRA with the BioProject number PRJNA1231493. We have provided the processed read counts and scripts to analyze them to the following GitHub repository: https://github.com/Thatguy027/fog2manuscript.
Reagents
Strains:
**: **
Bacteria:
**: **
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Andersen EC Bloom JS Gerke JP Kruglyak L 2014227 A variant in the neuropeptide receptor npr-1 is a major determinant of Caenorhabditis elegans growth and physiology.P Lo S Genet 1021553-7390 e 1004156 e 100415610.1371/journal.pgen.100415624586193 PMC 3937155 · doi ↗ · pubmed ↗
- 2Andersen EC Rockman MV 202214 Natural genetic variation as a tool for discovery in Caenorhabditis nematodes.Genetics 22010016-673110.1093/genetics/iyab 15635134197 PMC 8733454 · doi ↗ · pubmed ↗
- 3Andersen EC Shimko TC Crissman JR Ghosh R Bloom JS Seidel HS Gerke JP Kruglyak L 2015313 A Powerful New Quantitative Genetics Platform, Combining Caenorhabditis elegans High-Throughput Fitness Assays with a Large Collection of Recombinant Strains.G 3 (Bethesda)5591192010.1534/g 3.115.01717825770127 PMC 4426375 · doi ↗ · pubmed ↗
- 4Ben-David E Burga A Kruglyak L 2017511 A maternal-effect selfish genetic element in Caenorhabditis elegans.Science 35663420036-80751051105510.1126/science.aan 062128495877 PMC 6251971 · doi ↗ · pubmed ↗
- 5Brenner S 197451 The genetics of Caenorhabditis elegans.Genetics 7710016-6731719410.1093/genetics/77.1.714366476 PMC 1213120 · doi ↗ · pubmed ↗
- 6Burga A Ben-David E Lemus Vergara T Boocock J Kruglyak L 2019618 Fast genetic mapping of complex traits in C. elegans using millions of individuals in bulk.Nat Commun 1012680268010.1038/s 41467-019-10636-931213597 PMC 6582151 · doi ↗ · pubmed ↗
- 7C. elegans Sequencing Consortium. 19981211 Genome sequence of the nematode C. elegans: a platform for investigating biology.Science 28253960036-80752012201810.1126/science.282.5396.20129851916 · doi ↗ · pubmed ↗
- 8Chou HT Valencia F Alexander JC Bell AD Deb D Pollard DA Paaby AB 202413 Diversification of small RNA pathways underlies germline RNA interference incompetence in wild Caenorhabditis elegans strains.Genetics 22610016-673110.1093/genetics/iyad 19137865119 PMC 10763538 · doi ↗ · pubmed ↗