Popcorn: prediction of short coding and noncoding genomic sequences in prokaryotes
Alison Kyrouz, Lian Liu, Lixin Qin, Brian Tjaden

TL;DR
Popcorn is a machine learning tool that helps identify whether short prokaryotic genomic sequences are coding or noncoding.
Contribution
Popcorn introduces a novel machine learning method to distinguish coding and noncoding sequences in prokaryotes.
Findings
Popcorn effectively distinguishes coding sORFs from noncoding RNAs in prokaryotic sequences.
The method is accurate in identifying small transcripts that may encode novel proteins or regulatory RNAs.
Abstract
The most challenging prokaryotic genes to identify often correspond to short ORFs (sORFs) encoding small proteins or to noncoding RNAs. RNA-seq experiments commonly evince small transcripts that do not correspond to annotated genes and are candidates for novel coding sORFs or small regulatory RNAs, but it can be difficult to accurately assess whether the numerous small transcripts are coding or not. We present Popcorn (PrOkaryotic Prediction of Coding OR Noncoding), a novel machine learning method for determining whether prokaryotic sequences are coding or noncoding. We find that Popcorn is effective in distinguishing coding from noncoding sequences, including coding sORFs and noncoding RNAs. Freely available for use on the web at https://cs.wellesley.edu/∼btjaden/Popcorn. Source code available at https://github.com/btjaden/Popcorn and https://doi.org/10.5281/zenodo.15120075.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
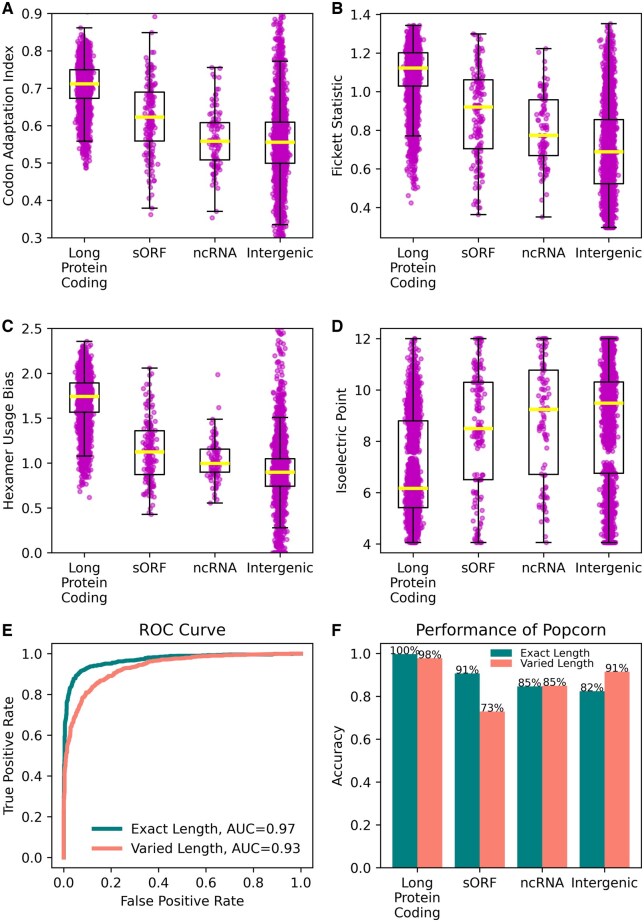 Figure 1
Figure 1- —National Institutes of Health10.13039/100000002
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Phylogenetic Studies · RNA and protein synthesis mechanisms · Machine Learning in Bioinformatics
1 Introduction
Small proteins, also called microproteins or small peptides, are commonly defined as proteins containing up to 50 amino acids. The short ORFs (sORFs) encoding small proteins are generally overlooked by popular gene annotation systems owing to the challenges in accurately identifying short genes (Fuchs and Engelmann 2023). Yet, small proteins are prevalent in eukaryotes (Schlesinger and Elsasser 2022), prokaryotes (Duan et al. 2024), and phages (Fremin et al. 2022), and have important and varied functional roles, such as in regulation (Altieri et al. 2016), sporulation (Schmalisch et al. 2010), and as components of toxin-antitoxin systems (Sonika et al. 2023). Among prokaryotes, it has been observed that small proteins occur at a proportionally higher rate in archaea than in bacteria (Duan et al. 2024). In Escherichia coli (E. coli), perhaps the best studied prokaryote, more than 150 small proteins have been characterized and it is hypothesized that many more remain to be identified (Hemm et al. 2020, Gray et al. 2022).
Prokaryotic RNA-seq experiments often detect hundreds or even thousands of short transcripts that do not correspond to previously annotated genes (Sharma et al. 2010, Thomason et al. 2015). It remains a major open question as to what extent these transcripts correspond to previously uncharacterized genes, including new sRNAs (small noncoding regulatory RNAs) or protein encoding sORFs, or instead reflect noise and promiscuous transcription (Wade and Grainger 2014). While experimental approaches, such as ribosome profiling (Vazquez-Laslop et al. 2022) and mass spectrometry (Ahrens et al. 2022), can be effective in identifying small proteins, they do not necessarily scale with the pervasiveness of RNA-seq experiments and their detection of novel short transcripts. Thus, there is value in having computational methods, which can be more efficient than experimental methods, for determining whether novel transcripts observed from RNA-seq experiments are likely to represent protein encoding sORFs.
Owing to the importance of identifying which short transcripts are coding or not in the current age of abundant RNA-seq data, a number of computational tools have been developed for distinguishing coding sequences from noncoding sequences (Washietl et al. 2011, Zhu and Gribskov 2019, Zhang et al. 2021). The vast majority of tools for predicting protein encoding sORFs have been designed for eukaryotic data [reviewed in Schlesinger and Elsasser (2022)]. For prokaryotes, studies aimed at characterizing coding sORFs have focused primarily on well conserved families of small proteins. For instance, Sberro et al. (2019) search for instances of small proteins from conserved families in the human microbiome, and GMSC-mapper searches sequences against its large catalog, GMSC, of microbial sORF families in order to identify new members of the families (Duan et al. 2024). One of the limitations of these methods is that they are ineffective for sORFs that are not well conserved, which is common among these short sequences.
Here, we present a new method, Popcorn (PrOkaryotic Prediction of Coding OR Noncoding), for predicting whether prokaryotic sequences are coding or noncoding. Our method makes a number of contributions. First, it uses a neural network trained exclusively on prokaryotic data, including a large number of sORFs coding for small proteins (as examples of coding sequences) and a large number of sRNAs (as examples of noncoding sequences). For RNA-seq experiments that evince small transcripts that do not correspond to annotated genes, these training data enable Popcorn to effectively distinguish coding and noncoding sequences for common small gene families in prokaryotes. Second, Popcorn does not require sequences to be conserved, which is advantageous since many small genes both coding and noncoding are not well conserved. Third, for effective prediction, Popcorn does not require that a sequence begin and end precisely at transcription or translation initiation and termination points. A number of tools for eukaryotic data are trained and tested on sequences corresponding to coding ORFs, i.e. the translated portion of a gene beginning at a start codon and ending at a stop codon. In practice, transcripts observed from bulk RNA-seq experiments are not so precise and may represent only part of a gene owing, e.g. to processing or degradation, or may extend beyond the coding sequence because they include UTRs (untranslated regions). To address this issue, Popcorn was trained both on truncated sequences and on sequences extended upstream and downstream of a gene boundary. Fourth, to enable ease of use and reproducibility, Popcorn is available via a webserver at https://cs.wellesley.edu/∼btjaden/Popcorn.
2 Results
We gathered a large set of prokaryotic genomic sequences corresponding to small genes, such as protein coding sORFs and ncRNAs, and we examined a variety of sequence features that may have predictive power in distinguishing the sequences as coding or noncoding (Supplementary Material). We investigated the distributions of features for four different types of sequences: long protein coding, sORFs, ncRNAs, and intergenic. The four types of sequences reflect, respectively, long coding sequences, short coding sequences, short noncoding sequences, and long noncoding sequences. We observed that the features indeed had different distributions for different types of sequences (Fig. 1A–D). For example, we found that the codon adaptation index (Fig. 1A), Fickett statistic (Fig. 1B), and hexamer usage bias (Fig. 1C) were higher for coding sequences (long protein coding and sORFs) and lower for noncoding sequences (ncRNAs and intergenic), whereas the isoelectric point (Fig. 1D) was lower for coding sequences (long protein coding and sORFs) and higher for noncoding sequences (ncRNAs and intergenic). Thus, these features contain some discriminatory properties and might reasonably be combined to discern coding from noncoding sequences.
For four of the features, (A) Codon Adaptation Index, (B) Fickett Statistic, (C) Hexamer Usage Bias, and (D) Isoelectric Point, the distribution of the feature’s values is shown for each of four types of genomic sequences: long protein coding (coding for proteins with >50 amino acids), sORFs (coding for proteins with no >50 amino acids), ncRNAs (noncoding RNA genes, e.g. sRNAs, that do not correspond to “housekeeping” RNAs such as rRNAs and tRNAs), and noncoding intergenic sequences. Overlayed on each distribution is a box and whisker plot, where the horizontal line indicates the median of the distribution, the top and bottom of the box indicate the first and third quartiles of the distribution, and the whiskers extend from the box by 1.5 times the inter-quartile range. (E) the receiver operating characteristic (ROC) curve illustrating the performance of the trained neural network model on the testing data, on both exact length sequences and varied length sequences. (F) the performance of the neural network model on testing data as measured by accuracy on both exact length sequences and varied length sequences for four types of genomic sequences: long protein coding, sORFs, ncRNAs, and intergenic sequences.
Motivated by these results, we trained a neural network that integrates the different features (Supplementary Material). We then explored to what extent each feature contributes to the neural network’s output (Tjaden and Tjaden 2023). We calculated Shapley values to explain the neural network’s reliance on each feature (Lundberg and Lee 2017). We observed that some features, like the Fickett statistic, contribute more to the neural network’s inference whereas other features, such as the GC content and isoelectric point, contribute much less (Supplementary Fig. S1).
We evaluated the performance of our trained neural network model on testing data from prokaryotic genomes whose genus was not represented in the training data. We considered the AUC (area under the ROC curve) for both exact length sequences and varied length sequences that may be truncated and/or contain sequence upstream and downstream of the gene. In general, the model showed strong performance in characterizing coding and noncoding sequences, with AUCs of 0.97 and 0.93 for exact length sequences and varied length sequences, respectively (Fig. 1E). We also examined how the model performed on different types of sequences, namely on short (ncRNA and sORF) and long (intergenic and long protein coding) noncoding and coding sequences. For exact length sequences, the model correctly identified the sequence as coding or noncoding between 82% (intergenic) and 100% (long protein coding) of the time (Fig. 1F). For varied length sequences, the model correctly identified the sequence as coding or noncoding between 73% (sORF) and 98% (long protein coding) of the time (Fig. 1F).
The somewhat lower performance (73%) on varied length sORF sequences suggests that, for these sequences, the model identifies them as coding more accurately when the sequence more precisely reflects the sORF as opposed to noncoding regions upstream/downstream or truncated portions of the sORF. To test this hypothesis, we explored how the performance of the model changes for sequences that include different length regions upstream/downstream or that are truncated by different amounts. For noncoding sequences, using the precise boundary of the sequence has little effect on identifying the sequence as noncoding (Supplementary Fig. S2). However, for coding sequences, the model performs best when the entire coding region is included and the performance drops as smaller portions of the coding sequence are considered (Supplementary Fig. S2). It is perhaps unsurprising that the performance of the model is compromised most for sORFs when the integrity of the sequence length is varied since these are the shortest types of sequences, averaging 127 nucleotides in length.
Arguably the most challenging family of genes to characterize as coding or noncoding are dual-function sRNAs that act both as base-pairing regulatory RNAs and as peptide encoding mRNAs. A recent review defines 11 such dual-function sRNAs that have been identified throughout prokaryotes to date (Schnoor et al. 2024). To assess the model’s ability to characterize these difficult genes, for each of the 11 dual-function sRNAs we tested the model on the entire RNA sequence as well as on the peptide encoding portion of the sequence. We found that the model identifies all 11 peptide encoding regions as coding regions and identifies 7 of the 11 complete RNA sequences as noncoding (Supplementary Fig. S3), suggesting that the model performs well but not perfectly on these challenging genes.
3 Discussion
Popcorn is a tool for determining whether prokaryotic genomic sequences are protein coding or noncoding. Popcorn uses a neural network trained on a large set of prokaryotic sequences, including short coding and noncoding genes. A common use case for Popcorn arises following an RNA-seq experiment where small transcripts are observed that do not correspond to annotated genes. Popcorn can distinguish protein coding sORFs from other transcripts such as noncoding RNAs, which are some of the most challenging prokaryotic genes to identify owing to their small size and, often, their lack of conservation in other species. Popcorn is available to use via a user-friendly web interface at https://cs.wellesley.edu/∼btjaden/Popcorn.
Supplementary Material
btaf250_Supplementary_Data
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Ahrens CH , Wade JT, Champion MM et al A practical guide to small protein discovery and characterization using mass spectrometry. J Bacteriol 2022;204:e 0035321.34748388 10.1128/jb.00353-21PMC 8765459 · doi ↗ · pubmed ↗
- 2Altieri AS , Ladner JE, Li Z et al A small protein inhibits proliferating cell nuclear antigen by breaking the DNA clamp. Nucleic Acids Res 2016;44:6232–41.27141962 10.1093/nar/gkw 351PMC 5181682 · doi ↗ · pubmed ↗
- 3Duan Y , Santos-Júnior CD, Schmidt TS et al A catalog of small proteins from the global microbiome. Nat Commun 2024;15:7563.39214983 10.1038/s 41467-024-51894-6PMC 11364881 · doi ↗ · pubmed ↗
- 4Fremin BJ , Bhatt AS, Kyrpides NC et al; Global Phage Small Open Reading Frame (GP-Sm ORF) Consortium. Thousands of small, novel genes predicted in global phage genomes. Cell Rep 2022;39:110984.35732113 10.1016/j.celrep.2022.110984 PMC 9254267 · doi ↗ · pubmed ↗
- 5Fuchs S , Engelmann S. Small proteins in bacteria—big challenges in prediction and identification. Proteomics, 2023;23:e 2200421.37609810 10.1002/pmic.202200421 · doi ↗ · pubmed ↗
- 6Gray T , Storz G, Papenfort K. Small proteins; big questions. J Bacteriol 2022;204:e 0034121.34309401 10.1128/JB.00341-21PMC 8765408 · doi ↗ · pubmed ↗
- 7Hemm MR , Weaver J, Storz G. Escherichia coli small proteome. Eco Sal Plus 2020;9:10–1128.10.1128/ecosalplus.esp-0031-2019 PMC 721291932385980 · doi ↗ · pubmed ↗
- 8Lundberg SM , Lee SI. A unified approach to interpreting model predictions. Adv Neural Inform Process Syst 2017;30:4765–74.