The innate immune system: A double-edged sword
Ditte S. Andersen, Julien Colombani

TL;DR
The body's first line of defense can both fight and help cancer grow, as shown by new research on immune signaling and cancer genes.
Contribution
Two studies reveal how cancer cells use innate immune signals to grow unchecked.
Findings
Premalignant cells use Toll-NF-κB signaling to support tumor growth.
This signaling works together with the Ras oncogene to enable uncontrolled cell growth.
The findings highlight the dual role of the innate immune system in cancer.
Abstract
Innate immunity serves as a crucial surveillance framework, but can be exploited to facilitate tumor progression. Two new PLOS Biology studies independently show how premalignant cells can exploit Toll-NF-κB signaling, in concert with oncogenic Ras, to enable unchecked growth
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
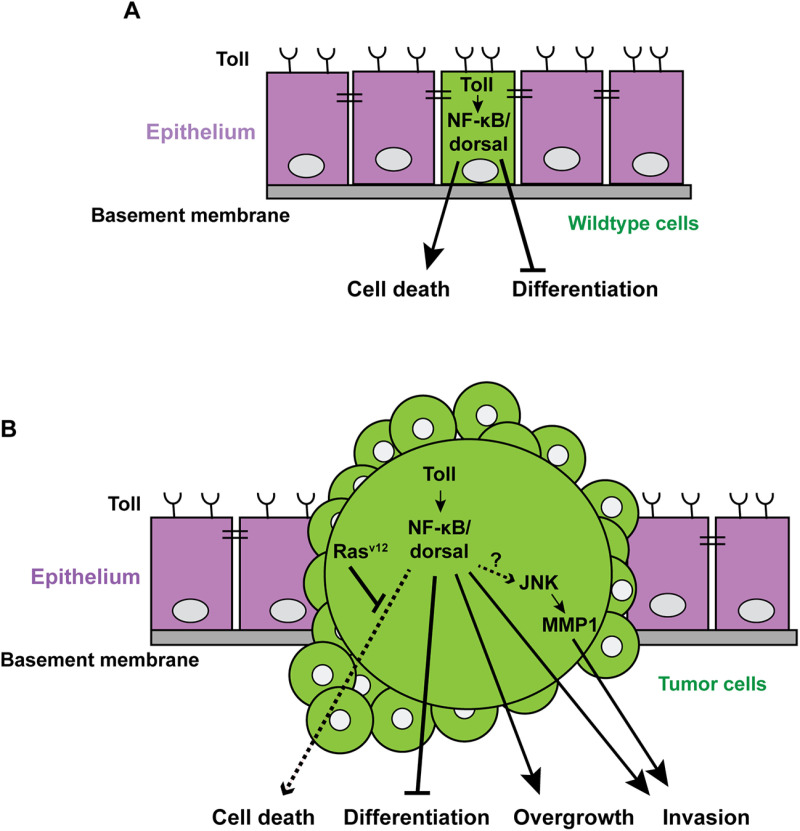 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsImmune Response and Inflammation · Immune cells in cancer · Neutrophil, Myeloperoxidase and Oxidative Mechanisms
Innate immunity serves as a global surveillance system, sensing external stimuli and assessing cellular fitness to initiate context-specific responses that combat infections and maintain tissue homeostasis. The Toll pathway, conserved from invertebrates to mammals, is a crucial component of the innate immune system. Its activation triggers a signaling cascade that leads to the translocation of the transcription factor, NF-κB (Dorsal and Dif in Drosophila), into the nucleus, driving the production of host defense peptides. Beyond its role in immunity, the Toll pathway has also been linked to cell competition - a process that eliminates less fit cells through apoptosis to uphold tissue homeostasis [1,2]. Notably, the proinflammatory properties of innate immune signals can be exploited by malignant cells to facilitate tumor progression. In Ras^v12^/scrib^-/-^ tumors, this is exemplified by their ability to hijack Tumor Necrosis Factor (TNF)/Eiger to enhance tumor growth and invasion [3,4]. The Ras^V12^/scrib^−/−^ tumor model has been widely utilized in Drosophila cancer research, as it exhibits a striking array of hallmarks reminiscent of human malignancies [5]. In this model, the combination of oncogenic Ras (Ras^V12^) expression with loss of cell polarity due to scribble loss-of-function results in malignant cells that are refractory to apoptosis, leading to the formation of aggressive, invasive tumors (Fig 1) [6,7]. Similarly, Toll-like receptor (TLR) signaling is frequently hyperactivated in malignant cells [8], although the mechanisms by which Toll pathway synergizes with cancer-driving mutations to promote tumor growth remain unclear. To explore this, two recent studies in PLOS Biology, Brutscher F. and colleagues and Dillard C. and colleagues, employed Drosophila Ras^V12^-driven epithelial tumor models to investigate how Toll-NF-κB signaling cooperates with oncogenic Ras to drive unrestricted growth and invasiveness [9,10].
Brutscher and colleagues show that while Toll-NF-κB pathway activation alone reduces tissue size, its activation in conjunction with oncogenic Ras^V12^ creates a potent oncogenic synergy, driving tissue overgrowth. Using RNA sequencing, they identified genes that are differentially expressed upon co-expression of Ras^V12^ and Dorsal (Dl) [10]. Their analyses reveal that Toll pathway activation suppresses differentiation markers; a finding they experimentally validate in vivo. Additionally, they show that although Toll-NF-κB signalling induces apoptosis, its activation alongside with Ras^V12^, which inhibits apoptosis, shifts the effect toward increased cell proliferation. Notably, despite upregulation of several JNK/AP-1 target genes, JNK is not required downstream of Dorsal to amplify Ras^V12^-driven tissue overgrowth.
Through RNA sequencing of Ras^v12^/scrib^−/−^ tumors, Dillard and colleagues identify several Toll pathway components as upregulated and demonstrate an autonomous requirement for Toll-NF-κB signaling to promote Ras^v12^/scrib^−/−^ tumor growth. Focusing on Dl, the authors examine the two alternatively spliced isoforms, DlA and DlB, both of which are upregulated in Ras^v12^/scrib^−/−^ tumors. Notably, these isoforms display an asymmetric distribution, with elevated expression in the posterior region of the tumor near the central nervous system and the highest levels in tumor cells actively invading neighbouring tissues. Furthermore, DlA and DlB display a mutually exclusive expression pattern with DlB being expressed in neurons within the tumor and DlB in non-neuronal cells surrounding these. Intriguingly, DlB expression shows a positive correlation with JNK activity, a key driver of Ras^v12^/scrib^−/−^ tumor invasiveness. Consistent with this, they show that silencing of Dl partially restores retinal differentiation, increases apoptosis and reduces tumor invasion. These findings raise the intriguing possibility that each isoform could serve a distinct function, with DlB driving JNK-mediated invasion and DlA supporting stemness and survival. Uncovering the mechanisms that regulate differential Dl splicing and how this contributes to tumorigenesis presents an interesting line of research.
Altogether, Brutscher F. and colleagues and Dillard C. and colleagues demonstrate that the Toll-NF-κB signaling cooperates with cancer-driving mutations to suppress differentiation, counteract apoptosis, and promote invasion [9,10]. One open question remains regarding how the innate immune system is activated in these tumors. Intriguingly, results from a recent study using a Yorkie-driven intestinal tumor model suggest that innate immunity may be triggered through interactions between the tumor and its microenvironment. Their findings shows that TNF/Eiger production from Yki^act^ tumors induces cell-competition-driven apoptosis in neighboring non-tumor cells. This, in turn, initiates an NF-κB-driven inflammatory response within the tumor, possibly mediated by damage-associated molecular patterns released from dying cells [11].
The role of the Toll-NF-κB signalling pathway in tumor growth parallels that of another innate immune signal, TNF, which can function as both a tumor promoter and suppressor. In Drosophila, the TNF homolog, Eiger, plays a key role in an antitumor surveillance system that facilitates the elimination of premalignant cells from peripheral tissues. However, like Toll-NF- κB signalling, Eiger/TNF can also be repurposed into a protumor signal with harmful consequences for the host. For instance, the invasive behaviour of Ras^V12^/scrib^−/−^ tumors is driven by Eiger/TNF, which activates JNK signalling and drives the expression of matrix metalloproteinase-1, promoting tumor invasion and progression [3,4].
The observed heterogeneity of NF-kB/Dl levels and activity mirrors patterns observed in mammalian cancers and raises important questions regarding the differential immune response of malignant cells. Understanding whether this heterogeneity stems from extrinsic signals from nearby cells in the microenvironment or intrinsic regulatory mechanisms, as well as how intratumoral NF-kB heterogeneity contributes to malignant transformation, presents an exciting avenue for future research.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Meyer SN, Amoyel M, Bergantiños C, de la Cova C, Schertel C, Basler K, et al. An ancient defense system eliminates unfit cells from developing tissues during cell competition. Science. 2014;346(6214):1258236. doi: 10.1126/science.1258236 25477468 PMC 5095928 · doi ↗ · pubmed ↗
- 2Alpar L, Bergantiños C, Johnston LA. Spatially restricted regulation of Spätzle/toll signaling during cell competition. Dev Cell. 2018;46(6):706–719.e 5. doi: 10.1016/j.devcel.2018.08.001 30146479 PMC 6156939 · doi ↗ · pubmed ↗
- 3Cordero JB, Macagno JP, Stefanatos RK, Strathdee KE, Cagan RL, Vidal M. Oncogenic Ras diverts a host TNF tumor suppressor activity into tumor promoter. Dev Cell. 2010;18(6):999–1011. doi: 10.1016/j.devcel.2010.05.014 20627081 PMC 3175220 · doi ↗ · pubmed ↗
- 4Andersen DS, Colombani J, Palmerini V, Chakrabandhu K, Boone E, Röthlisberger M, et al. The Drosophila TNF receptor Grindelwald couples loss of cell polarity and neoplastic growth. Nature. 2015;522(7557):482–6. doi: 10.1038/nature 14298 25874673 · doi ↗ · pubmed ↗
- 5Bilder D, Ong K, Hsi T-C, Adiga K, Kim J. Tumour-host interactions through the lens of Drosophila. Nat Rev Cancer. 2021;21(11):687–700. doi: 10.1038/s 41568-021-00387-5 34389815 PMC 8669834 · doi ↗ · pubmed ↗
- 6Brumby AM, Richardson HE. Scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J. 2003;22(21):5769–79. doi: 10.1093/emboj/cdg 548 14592975 PMC 275405 · doi ↗ · pubmed ↗
- 7Pagliarini RA, Xu T. A genetic screen in Drosophila for metastatic behavior. Science. 2003;302(5648):1227–31. doi: 10.1126/science.1088474 14551319 · doi ↗ · pubmed ↗
- 8Maiorino L, Dassler-Plenker J, Sun L, Egeblad M. Innate immunity and cancer pathophysiology. Annual Review of Pathology. 2022;17:425–57.10.1146/annurev-pathmechdis-032221-115501 PMC 901218834788549 · doi ↗ · pubmed ↗