The complete genome sequence of the crayfish pathogen Candidatus Paracoxiella cheracis n.g. n.sp. provides insight into pathogenesis and the phylogeny of the Coxiellaceae family
Danielle J. Ingle, Calum J. Walsh, Genevieve R. Samuel, Ryan R. Wick, Nadav Davidovich, Eleonora Fiocchi, Louise M. Judd, Jennifer Elliman, Leigh Owens, Timothy P. Stinear, Andrea Basso, Tobia Pretto, Hayley J. Newton

TL;DR
Scientists discovered a new bacterial genus that infects crayfish and found it has a unique system for invading host cells.
Contribution
The paper introduces a new genus in the Coxiellaceae family and reveals its genome and unique intracellular mechanisms.
Findings
The genome of Candidatus Paracoxiella cheracis was fully sequenced using long-read metagenomics.
The organism encodes a functional Dot/Icm type 4 secretion system and unique effector proteins.
The findings provide insights into the evolution and pathogenesis of Coxiellaceae bacteria.
Abstract
The Coxiellaceae bacterial family, within the order Legionellales, is defined by a collection of poorly characterized obligate intracellular bacteria. The zoonotic pathogen and causative agent of human Q fever, Coxiella burnetii, represents the best-characterized member of this family. Coxiellaceae establish replicative niches within diverse host cells and rely on their host for survival, making them challenging to isolate and cultivate within a laboratory setting. Here, we describe a new genus within the Coxiellaceae family that has been previously shown to infect economically significant freshwater crayfish. Using culture-independent long-read metagenomics, we reconstructed the complete genome of this novel organism and demonstrate that the species previously referred to as Candidatus Coxiella cheraxi represents a novel genus within this family, herein denoted Candidatus Paracoxiella…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
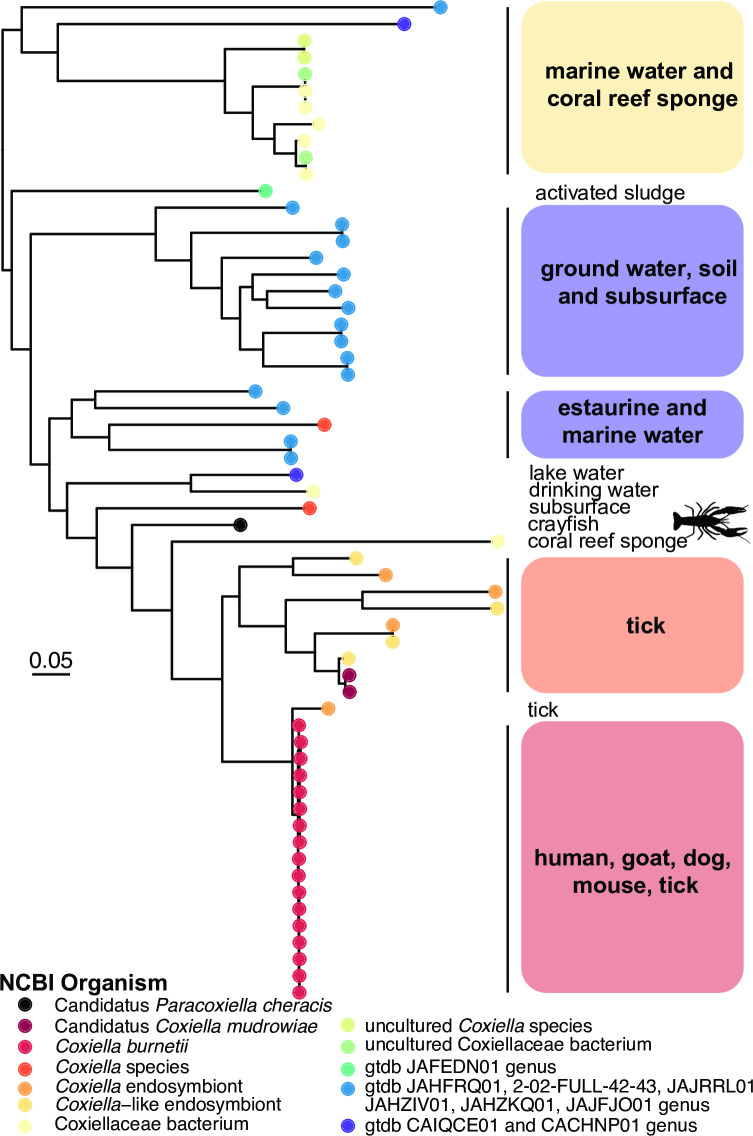 Fig 1
Fig 1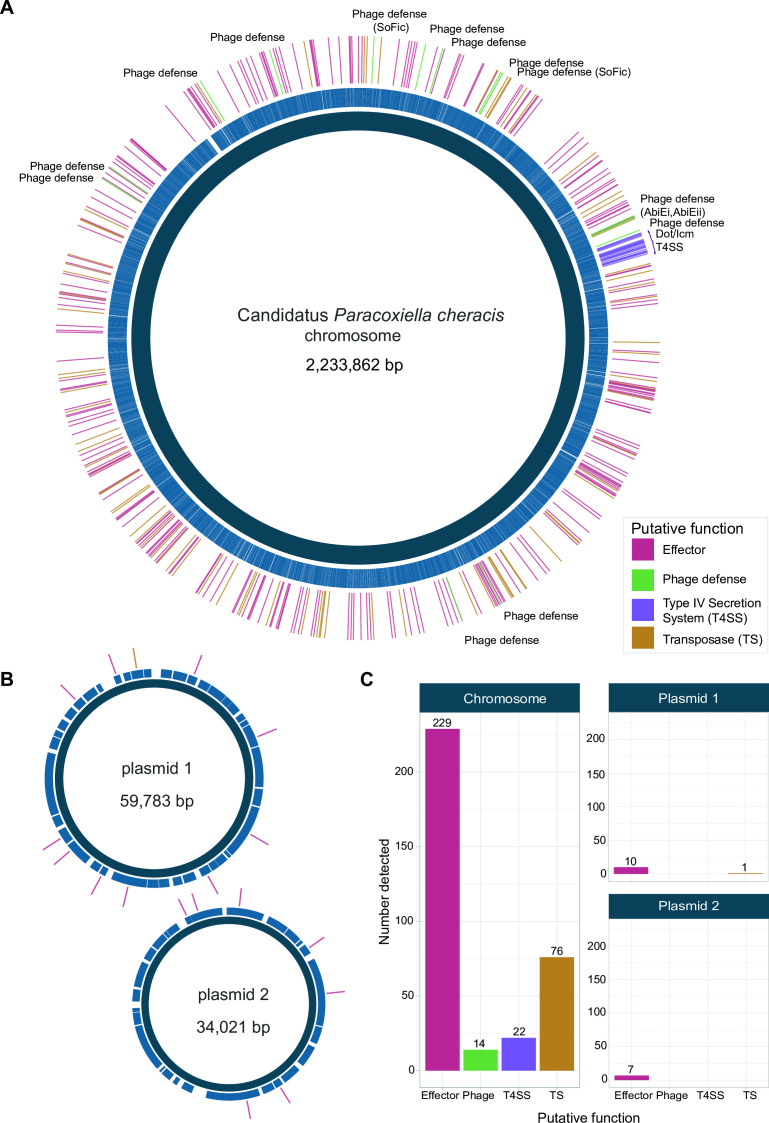 Fig 2
Fig 2 Fig 3
Fig 3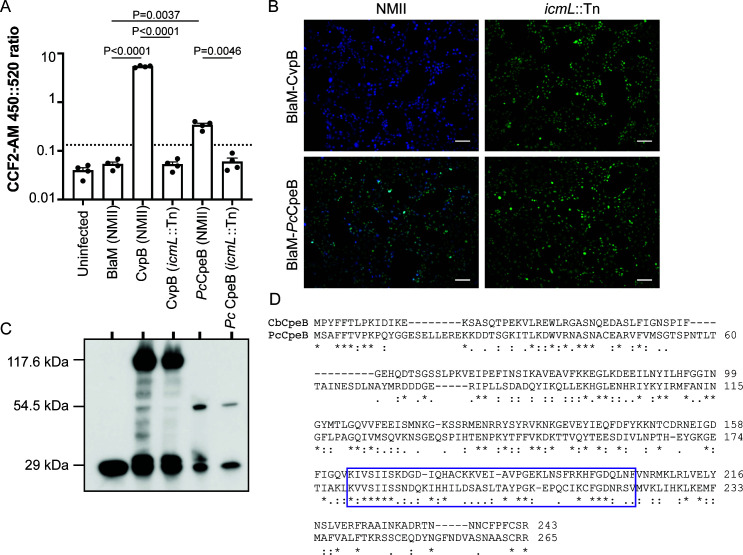 Fig 4
Fig 4| Candidatus | Identity (%) | Coverage (%) | Role of | |
|---|---|---|---|---|
| PBJCNB_00280 | CBUD_0487 | 26 | 22 | Experimentally shown to be a Dot/Icm substrate ( |
| PBJCNB_01005 | CBU_1863 (CvpE) | 40 | 78 | |
| PBJCNB_01155 | CBU_0201 (AnkC) | 84 | 95 | Early studies using |
| PBJCNB_01790 | CBUA_0013 (CpeB) | 29 | 90 | Plasmid-encoded |
| PBJCNB_05220 | CBU_0937 (CirC/MceB) | 57 | 100 | Identified as a DotF binding protein and demonstrated to be translocated by |
| PBJCNB_05510 | CBU_1079 | 76 | 97 | 5% translocation observed using the BlaM reporter in |
| PBJCNB_08410 | CBU_0513 (CinF) | 83 | 99 | Shown to be translocated as a BlaM-fusion protein by |
| PBJCNB_09120 | CBU_1425 | 74 | 100 | Identified as a DotF binding protein and demonstrated to be translocated by |
| PBJCNB_09430 | CBU_0469 | 61 | 95 | Low-level (1%) translocation of BlaM fusion protein in |
| PBJCNB_10235 | CBU_1566 | 79 | 99 | BlaM fusion protein was shown to be translocated by |
| PBJCNB_10940 | CBU_0175 | 82 | 100 | BlaM fusion protein translocated by |
| PBJCNB_10315 | CBU_1594 | 71 | 97 | BlaM fusion protein was shown to be translocated by |
- —National Health and Medical Research Councilhttp://dx.doi.org/10.13039/501100000925
- —National Health and Medical Research Councilhttp://dx.doi.org/10.13039/501100000925
- —National Health and Medical Research Councilhttp://dx.doi.org/10.13039/501100000925
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLegionella and Acanthamoeba research · Vibrio bacteria research studies · Mosquito-borne diseases and control
INTRODUCTION
Australian native freshwater redclaw crayfish, Cherax quadricarinatus, are bred for the commercial market. Aquaculture of these animals has led to the observation of infectious diseases that impact both wild and farmed C. quadricarinatus (reviewed in reference 1). In particular, a Rickettsial-like intracellular bacterium, designated TO-98, isolated from infected crayfish was recognized as the likely causative agent of mass mortality in farmed redclaw crayfish in the early 1990s (2, 3). Subsequent investigations were able to recapitulate disease in experimentally infected crayfish and demonstrate that TO-98 causes a lethal infection in redclaw crayfish (4). This study conducted 16S rRNA sequence analysis and revealed that TO-98 was closely related to the zoonotic human pathogen Coxiella burnetii, coining the name Candidatus Coxiella cheraxi (4).
The initial observation that this pathogen is a member of the Coxiellaceae family has been confirmed by examination of additional DNA sequences, including comparison of specific gene sequences (5) and MinION sequencing data showing fragments of DNA with closest similarity to C. burnetii and Coxiella-like endosymbionts (C-LEs) (6). However, this fragmented genomic data do not allow complete and comprehensive understanding of the relationship between C. burnetii and Candidatus C. cheraxi.
C. burnetii, the causative agent of human Q fever, has a large reservoir in domesticated ruminants (7). C. burnetii is a Gammaproteobacteria within the order Legionellales and family Coxiellaceae. While the order Legionellales is best known for the two major human pathogens, Legionella pneumophila and C. burnetii, these bacteria are distantly related, and the order is poorly characterized, including over 450 uncultured genera (8). The diversity of Legionellales has been revealed by increased sampling of different environments and ecological surveys employing metagenomic techniques that have demonstrated the presence of Legionellales within arthropods, amoeba, and aquatic environments (reviewed in reference 9). Most of these newly discovered Legionellales are likely nonpathogenic bacteria that have evolved mutualistic relationships with non-vertebrate hosts. However, recent data suggest that some poorly characterized Legionellales harbor the potential to be zoonotic pathogens and/or pathogenic to important animal species. This includes a recently identified Coxiella species found in the placenta of fur seals (10).
Essential to the ability of C. burnetii and L. pneumophila to replicate intracellularly and cause disease is the Dot/Icm type IVB secretion system (T4SS) (11–14). This multiprotein apparatus is responsible for the delivery of a large cohort of effector proteins into the host cell where they modulate a range of cellular functions and pathways (reviewed in reference 15). Dot/Icm-deficient L. pneumophila are rapidly destroyed via lysosomal degradation as many of the effectors act to remodel the phagosome and block endocytic maturation (reviewed in reference 16). In contrast, internalized C. burnetii is trafficked through the endocytic pathway before the lysosomal environment triggers the activation of the Dot/Icm system (17, 18). Dot/Icm-deficient C. burnetii are not destroyed by the lysosome, but they are also incapable of intracellular replication (11, 12). Interestingly, comparative genomic studies show that the Dot/Icm system is lost or pseudogenized in CL-Es, indicating its association with pathogenesis (19).
Despite L. pneumophila and C. burnetii relying on the same key virulence factor, the cohort of effector proteins translocated by this secretion system is pathogen specific. This genetic divergence is represented by phenotypic divergence through the establishment of distinct intracellular niches. The biochemical and functional characterization of these effector proteins is an active area of research as these novel proteins facilitate insight into both mechanisms of pathogenesis and novel strategies to manipulate eukaryotic cell biology.
Here, we report the complete genome sequence of Candidatus C. cheraxi, revealing a more distant relationship to C. burnetii than expected, notably that Candidatus C. cheraxi represents a novel genus within Coxiellaceae. We propose to rename this crayfish pathogen Candidatus Paracoxiella cheracis. Interestingly, we report that this novel Coxiellaceae family member possesses a Dot/Icm T4SS most closely associated with C. burnetii. In silico analysis allowed us to identify a large cohort of putative effectors of this secretion system, with only 12 out of 238 predicted effectors sharing identity with known C. burnetii T4SS effectors. We have been able to demonstrate the translocation of the Candidatus P. cheracis CpeB homolog via the C. burnetii Dot/Icm system. This study provides additional insight into the poorly characterized Coxiellaceae family and demonstrates that the Dot/Icm secretion system is a common tool used by these bacteria to communicate with their host. It also demonstrates the utility of long-read sequencing approaches for generating a complete metagenome-assembled genome for an obligate intracellular pathogen.
MATERIALS AND METHODS
Collection and preparation of genetic material
Samples of hepatopancreas from infected redclaw crayfish were collected during an outbreak of crayfish rickettsiosis, which occurred after the crayfish were imported from Australia to Israel in 2019 (20). Briefly, 10,000 juvenile redclaw crayfish (2–3 weeks old) were imported to Israel and quarantined in a facility. From April to July, juvenile mortality sharply increased despite the absence of clinical signs or gross lesions. Specimens collected for histological and molecular evaluations were sent to the Istituto Zooprofilattico Sperimentale delle Venezie in Legnaro, Italy, and revealed a severe infection by Candidatus C. cheraxi. DNA was extracted from these samples, as previously described (20), and sent to Monash University, Australia, for further analysis.
Long-read sequencing and genome assembly
Metagenomic sequencing of extracted DNA was performed on two separate sequencing runs. The first run was performed on an Oxford Nanopore Technologies (ONT) MinION, while the second run employed a PromethION. Both runs were carried out on R10.4.1 flow cells. Basecalling and quality filtering were performed by Dorado (version 0.5.0) using models [email protected] and [email protected], respectively. Reads shorter than 1 kbp were discarded. Kraken2 (21) was used for the initial read-level classification of metagenomic data.
Initial assemblies were generated using Flye (22), MetaMDBG (23), Miniasm (24), and Raven (25), each of which produced a single, continuous sequence representing the bacterial chromosome, and most of them contained two medium-sized plasmids at a similar read depth to the chromosome. For each assembly, the length-filtered reads were mapped to the chromosome and plasmids using Minimap2 (26), and the mapped reads were retained for downstream analysis. The chromosomal and plasmid reads were then downsampled to 500× coverage using Filtlong (github.com/rrwick/Filtlong). The shortest mapped read was 6,961 bp, and the mapped read N50 was 10,724 bp. Mapped reads were combined into a single high-quality assembly with Trycycler (27) incorporating 24 separate assemblies from six assemblers: Canu (28), Flye (22), Miniasm (24), NECAT (29), NextDenovo (30), and Raven (25). The final complete genome assembly was annotated with Bakta version 1.9.1 (31). Reads that mapped to the assembled chromosome were also classified with Kraken2. Full details of the individual steps for quality controls, kraken assignment, and genome assembly are available in File S1.
Phylogeny of Coxiellaceae family
The Genome Taxonomy Database Toolkit (GTDB-Tk) (32) classified this genome as Coxiellaceae. Representative isolates from the order Coxiellales (gtdb.ecogenomic.org/tree?r = o__Coxiellales) were used in comparative analyses to infer the relative location of Candidatus P. cheracis in the order. Given the large number of Coxiellales genomes in the GTDB database, the majority of which belong to the clinically important species Coxiella burnetii, the data set was first dereplicated by Assembly Dereplicator (github.com/rrwick/Assembly-Dereplicator) to a Mash distance of 0.001, resulting in a representative set of 60 genomes capturing the available diversity of the Order. GTDB-Tk (33), a marker gene-based tool for taxonomic classification and phylogenetic placement of prokaryotic genomes based on the Genome Taxonomy Database taxonomy system (34), was used to infer a phylogeny for this genome and the representative 60 genomes and to taxonomically classify our long-read assembly. Briefly, GTDB-Tk creates a protein multiple sequence alignment of bacterial marker genes in the genomes and uses FastTree to infer a phylogeny with the WAG-GAMA model as default. The final protein alignment was 5,035 amino acids in length.
Investigation of the complete genome for features of interest
The genome content of Candidatus P. cheracis was explored using publicly available tools. First, known antimicrobial resistance (AMR) determinants were screened with abritAMR (version 1.0.14) with the AMRfinderPlus database (v2022-08-09.1) with no species flag and default parameters (35). Mob-typer, which is part of Mob-Suite, was used to identify plasmid replicons and mob genes with default parameters (36, 37). Phage defense mechanisms were identified with PADLOC version 2 using the annotated gff file as input and default parameters (38).
Putative Dot/Icm effectors were screened in the complete genome with Bastion4 (39) and T4Sepp (40) tools. The predictions were compared, with hits for putative IS elements not included in further analysis.
Components of the T4SS were identified with SECRET4 (41). T4SSs were also identified in selected, publicly available genomes that have been established to have functional T4SSs. These included two C. burnetii genomes as the type strains for the species (RSA493 accession: AE016828.3 and Dugway accession: CP000733.1), two genomes from the Legionella genus, Legionella pneumophila subsp. pneumophila strain Philadelphia 1 (accession: AE017354.1) and Legionella longbeachae NSW150 (accession: NC_013861.1) (42) and the Candidatus Rickettsiella viridis Ap-RA04 genome (accession: AP018005.1). We also screened representative isolates with complete genomes from Coxiella like-endosymbionts (accessions: CP021379.1, CP011126.1, CP033868.1, and CP064834.1) with SECRET4 confirming the reported lack of a complete T4SS in these genomes (43).
Protein identity between the six genomes was undertaken with blastp using the Dot/Icm proteins from C. burnetii RSA493 (accession: AE016828.3) as a query sequence. For the three Coxiella genomes, the output was filtered to the top hit for each with a bitscore >100. A modified approach was undertaken for the two Legionella and Candidatus Rickettsiella viridis genomes due to the lower sequence similarity. For these three genomes, the results were filtered to the loci that encode the T4SS as identified by coordinates from SECRET4, and the hits with the highest bitscore for each protein were included.
Visualization of the Candidatus P. cheracis genome with features of interest including phage defense, T4SS, and putative Dot/Icm effectors was done in BRICK (github.com/esteinig/brick). The phylogenetic tree was visualized in R using ape (44) and ggtree (45). The R package gggenomes was used to plot the T4SS (github.com/thackl/gggenomes), and pheatmap was used to visualize the protein identity (github.com/raivokolde/pheatmap).
Cultivation of bacterial and tissue culture cells
C. burnetii Nine Mile phase II (NMII) strain RSA439 clone 4 was axenically cultivated in liquid ACCM-2 or ACCM-2 agarose at 37°C in 5% CO_2_ and 2.5% O_2_ as previously described (46). When required, chloramphenicol was added to C. burnetii cultures at 3 µg/mL. HeLa CCL2 cells (ATCC, VA, USA) were maintained in Dulbecco’s modified Eagle’s media supplemented with 10% heat-inactivated fetal bovine serum at 37°C in 5% CO_2_.
Construction of C. burnetii translocation reporter strains
The open reading frame encoding for the Candidatus P. cheracis CpeB homolog (PcCpeB) was synthesized by IDT and cloned into pJB-CAT:BlaM (47) using BamHI and XhoI restriction enzymes. The resulting pBlaM-PcCpeB was introduced into C. burnetii strains using a standard electroporation protocol (11). Following the selection of transformants, the expression of BlaM fusion proteins was confirmed using an anti-BlaM (1:1,500, QED Biosciences). C. burnetii strains carrying either pJB-CAT:BlaM or pJB-CAT:BlaM:CBU0021 were used as negative and positive translocation controls (48).
BlaM reporter translocation assay
Translocation assays were conducted as previously reported (17, 49). HeLa CCL2 cells were seeded in 96-well black, clear bottom tissue culture plates at a density of 10^4^ per well. Approximately 24 h later, cell monolayers were infected with C. burnetii strains at an MOI of 300. Following a 22 h incubation, wells were loaded with CCF2-AM substrate from the LiveBLAzer FRET loading kit (Thermo Fisher Scientific) and incubated at room temperature for 2 h. Wells were excited at 415 nm, and the emission at 450 nm (blue) and 520 nm (green) was collected using a Clariostar fluorescence plate reader. Representative images were acquired using an Invitrogen EVOS FL Imaging System, images taken at 10× objective.
RESULTS
A putative new genus in the Coxiellaceae family
Here, we describe the complete genome of a putative new genus within Coxiellaceae, Candidatus C. cheraxi, which is the likely causative agent of mass mortality events in C. quadricarinatus. The Candidatus C. cheraxi genome consisted of one chromosome (2,233,862 bp) and two smaller plasmids (59,783 and 34,021 bp). The chromosome is similar in size to previously sequenced C. burnetii isolates ranging from approximately 1.9–2.2 Mb (50). To confirm that this organism is related to the original Candidatus C. cheraxi TO-98 organism, the 21 available sequence reads from TO-98 (6) were mapped to this genome with an average identity of 89%. This high level of similarity, even with the TO-98 data set produced using older minION chemistry (R9.4.1), provides confidence that this genome is representative of the same species as TO-98.
The Candidatus C. cheraxi genome was classified as Coxiellaceae by GTDB-Tk (32), meaning that the novel genome could be classified to the family level but no further, suggesting that this genome represents a novel genus, supported by a relative evolutionary divergence value of 0.79373 (32). Based on these data, we propose to consider this Coxiellaceae as a novel genus: Candidatus Paracoxiella cheracis, also adopting the adjusted genus nomenclature (51).
A phylogenetic tree inferred from bacteria-specific marker genes placed Candidatus P. cheracis near the genomes of other Coxiellaceae bacteria (Fig. 1) isolated from similar aquatic ecological niches and distant to C. burnetii (largely associated with mammalian hosts) and the C-LEs that are associated with ticks as hosts (Fig. 1). Interestingly, Kraken2 classification of the chromosomal reads using a GTDB-based database identified multiple bacterial families in the read set (Files S1 and S2). These include 10.6% Alteromonadaceae, 4.1% Vibrionaceae, 3.7% Enterobacteriaceae, 2.9% Burkholderiaceae, 2.5% Legionellaceae, 2.3% Moraxellaceae, and only 8.5% Coxiellaceae (File S2), which are all members of the Gammaproteobacteria class. This demonstrates that Kraken2 results for an isolate in a novel taxon not represented in the database can be misleading and may falsely suggest a mixed sample. However, the assembly of the long-read data successfully recovered the bacterial chromosome and two plasmids.
Phylogeny of Coxiellales order demonstrating relationship to Candidatus P. cheracis genome. The inferred evolution of the Candidatus P. cheracis genome in representative isolates from Coxiellales order (gtdb.ecogenomic.org/tree?r = o__Coxiellales). The phylogeny was inferred using GTDB-Tk, a marker gene-based method of 120 domain-specific marker genes for bacteria (33). The tips of the tree are colored by the NCBI organism for the Coxiellales lineages and additional GTDB lineages at the genus level. The branch lengths represent the expected substitutions per site of the protein alignment of 5,035 amino acids. The associated hosts or sources from where the samples were collected are shown to the right of the tree. The Candidatus P. cheracis genome is indicated in black and by the crayfish icon (sourced from https://www.phylopic.org/nodes/aa251b9d-1cb9-4327-929a-d56ef1bc8f82/crayfishes-silhouettes).
The complete genome of Candidatus P. cheracis was explored for antimicrobial resistance, phage defense mechanisms, the Dot/Icm T4SS, and putative effectors of this system (Fig. 2). No known AMR mechanisms were identified, consistent with the AMR profiles of C. burnetii genomes. No replicon or mob genes were detected in the two plasmids. Furthermore, these two plasmid sequences had no similarity to publicly available data on NCBI. Several phage defense systems were identified on the chromosome in the novel genome. Two SoFic defense systems and the AbiE system, comprising AbiEi and AbiEii components, were identified in the genome. The remaining 10 defense systems identified were from the curated PADLOC database. Multiple putative effectors were identified (expanded upon in predicted substrates of the C. cheraxi Dot/Icm secretion system), in addition to many copies and variants of a transposase that shares identity with a transposase from a symbiont of Solemya velum, the Atlantic awning clam (accession: JRAA00000000) (52).
Characterization of the Candidatus P. cheracis genome. The complete genome showing (A) the chromosome and (B) two plasmids. The middle ring shows the coding sequence on the chromosome and plasmids. Genes of interest are shown in the outer ring with colors indicating broad function (T4SS, multicopy transposase (TS), phage defense, and putative effector). The total number of these four functional classes on each genetic element is shown on panel C.
Detection and analysis of the Dot/Icm locus encoded by Candidatus P. cheracis
The Dot/Icm system was detected in the Candidatus P. cheracis genome, suggesting that the isolate has a functional T4SS. The site of integration into the chromosome and gene arrangement were similar to the two isolates of C. burnetii examined here (Fig. 3A). The two C. burnetii genomes have additional genes between icmT and icmQ, some of which have been shown to be effector proteins (53), which are lacking in the Candidatus C. cheraxi genome. Additionally, icmT and icmS were encoded in the reverse orientation in the Candidatus P. cheracis genome compared to the two C. burnetii genomes. These two genes were in the same orientation as the two Legionella and Candidatus R. viridis genomes despite the Dot/Icm system being fragmented into multiple regions in these three genomes. Of note, the protein similarity between 21 genes of the Dot/Icm system differed within the six genomes by species and genus (Fig. 3B). The proteins from the C. burnetii Dugway isolate were homologous to the Dot/Icm proteins of C. burnetii RSA493 (>99% protein identity for all). In contrast, the protein identity of the Dot/Icm proteins in Candidatus P. cheracis varied between 52% (IcmG) and 86% (IcmB) relative to the C. burnetii RSA493 proteins. This was higher than the protein similarity of the Dot/Icm proteins from the two Legionella and the Candidatus R. viridis genomes, which ranged between 24% (IcmX in L. longbeachae) and 67% (DotB for Candidatus R. viridis). As previously reported, the Dot/Icm system was not detected in the three C-LE genomes.
Structure and protein identity of the T4SS in the Candidatus P. cheracis genome. (A) Characterization of the T4SS identified in Candidatus P. cheracis, Coxiella burnetii, Legionella pneumophila, Legionella longbeachae, and Candidatus Rickettsiella viridis. Genes encoding the T4SS are colored by function within the system. (B) Visualization of Dot/Icm protein identity relative to Coxiella burnetii RSA 493.
Predicted substrates of the Candidatus P. cheracis Dot/Icm secretion system
Given the conservation of genes encoding for the Dot/Icm apparatus, we hypothesized that Candidatus P. cheracis possesses a unique repertoire of putative T4SS substrates. To identify these proteins, two effector prediction pipelines, Bastion4 and T4SEpp, were employed. Bastion4, a bacterial secretion effector predictor for T4SS, is an ensemble effector predictor based on six distinct machine learning models (39). More recently, T4SEpp was developed using an integrated pipeline that incorporates homology-based predictions with machine learning models to generate a prediction score for the likelihood of a protein being a T4SS substrate (40). Combined, these tools predicted 355 putative Dot/Icm substrates within the Candidatus P. cheracis genome. Bastion4 predicted 296 effectors, and T4SEpp predicted 114, with 55 putative effectors predicted by both approaches (File S3). Bastion4 predicted 117 instances of IS elements as putative effectors. IS elements have not previously been reported as T4SS substrates; however, experimental evidence is required to determine whether these are false-positive results. Excluding IS elements, this approach identified a pool of 238 potential Dot/Icm effectors. Of particular interest, 66 putative effectors (25 predicted by both tools) have no significant similarity to protein sequences found in NCBI.
Candidatus P. cheracis CpeB, PcCpeB, can be translocated by the C. burnetii Dot/Icm system
Each of the predicted Dot/Icm effectors was examined for similarity to known Dot/Icm effectors of L. pneumophila and C. burnetii. No putative effectors showed similarity within the L. pneumophila effector cohort; however, 12 putative effectors have similarity to putative and experimentally characterized C. burnetii effectors (Table 1).
To test the feasibility of using C. burnetii as a surrogate bacterium to demonstrate Dot/Icm-dependent translocation of putative Candidatus P. cheracis effectors, the BlaM translocation reporter assay was employed. This assay is routinely used to demonstrate Dot/Icm substrate translocation into host cells and relies on bacterial delivery of a β-lactamase (BlaM) enzyme into the host cytosol where its activity can be measured by cleavage to the BlaM substrate CCF2-AM (11). Dot/Icm-dependent translocation of the Candidatus P. cheracis homolog of C. burnetii effector CpeB (PcCpeB) was confirmed (Fig. 4A and B). C. burnetii WT and Dot/Icm-deficient (*icmL::*Tn) strains were engineered to express BlaM alone (negative control), BlaM-CvpB, a well-characterized C. burnetii Dot/Icm effector (positive control) (48, 54, 65, 66) or BlaM-PcCpeB (Fig. 4C). HeLa cells were infected with these C. burnetii strains for 22 h before the addition of CCF2-AM and subsequent detection of fluorescence emission via both a fluorescence plate reader (Fig. 4A) and microscopy (Fig. 4B). BlaM-PcCpeB was translocated in a Dot/Icm-dependent manner. Interestingly, a comparison of the amino acid sequences of C. burnetii CpeB (CbCpeB) and PcCpeB showed only 29% identity; however, the region determined to mediate the interaction between CbCpeB and Rab11 shows greater conservation, potentially indicating functional similarity (purple box, Fig. 4D) (59).
Candidatus P. cheracis PcCpeB is a Dot/Icm substrate. HeLa cells were infected for 24 h with either wild-type (NMII) or Dot/Icm-deficient (icmL::Tn) C. burnetii expressing the indicated BlaM fusion proteins. (A) Change in the fluorescence emission ratio 450 nm::520 nm was measured as a readout of BlaM cleaving the CCF2-AM substrate (y-axis). The graph depicts this ratio from four independent experiments (circles) with the bar representing the mean and SEM ratio. The threshold for translocation positive was set to 2.5× the BlaM-alone negative control (dotted line), and Šidák’s multiple comparisons test was used to determine P-values compared to the BlaM and between NMII and icmL::Tn strains expressing the same fusion proteins. (B) Representative fluorescent images of infected HeLa cells demonstrate the translocation of CvpB (C. burnetii effector positive control) and PcCpeB by C. burnetii NMII but not C. burnetii icmL::Tn. Fluorescence intensity at 520 nm of uncleaved CCF2-AM is shown in green and cleaved CCF2-AM, at 450 nm is blue, scale bars represent 100 µm. (C) Immunoblot analysis of the indicated C. burnetii strains confirmed the expression of BlaM (29 kDa), BlaM-CvpB (117.6 kDa), and BlaM-PcCpeB (54.5 kDa). (D) ClustalOmega alignment showing the similarity between C. burnetii CpeB (CbCpeB) and Candidatus P. cheracis CpeB (PcCpeB). The two proteins share 29% identity (), with additional conserved (:) and semi-conserved (.) substitutions. The purple boxed area represents amino acids 164–204 that were previously shown to mediate the interaction of CbCpeB with Rab11a (59).*
DISCUSSION
In the absence of live samples of Candidatus P. cheracis n.g. n. sp., progressing our understanding of this novel crayfish pathogen is limited to the exploration of genetic material. This report of the complete genome sequence of this pathogen provides both an opportunity to expand our understanding of the evolutionary relationships between members of the family Coxiellaceae and formulate hypotheses regarding the host-pathogen interactions mediated by this organism. The absence of a viable specimen prevents the formal designation of this pathogen as a novel genus.
The phylogenetic placement, ANI to reference genomes, and relative evolutionary divergence of Candidatus P. cheracis provide evidence that this novel genome represents a new genus within the Coxiellaceae family and is more distantly related to both C. burnetii and C-LEs than previously appreciated. Of note, the Dot/Icm T4SS detected in Candidatus P. cheracis was found to have inserted into the chromosome near the same genes as in C. burnetii genomes. From this, we hypothesize that the Dot/Icm T4SS may have been acquired in the most recent common ancestor of both Candidatus P. cheracis, C. burnetii, and C-LEs. Moreover, the loss of the Dot/Icm T4SS in the C-LEs, which are associated with invertebrate hosts, suggests that the Dot/Icm T4SS did not confer a fitness advantage to warrant being maintained in C-LEs, which are maternally inherited in ticks (19).
The Dot/Icm T4SS has co-evolved in these different genetic backgrounds, resulting in the observed differences in protein identity between Candidatus P. cheracis and the two C. burnetii reference genomes, with the adaptation of the effectors to the different host niches. The observed differences in Dot/Icm protein identity between these genomes are greater than the average values recently reported for 58 Legionella species (67). However, the Dot/Icm components of Legionella with the greatest variation in protein identity were the same in the Candidatus P. cheracis (relative to C. burnetii), specifically IcmX, DotA, and IcmG/DotF, consistent with the exposure of these proteins to hosts (67). In contrast, IcmB and DotB had the highest protein identity, which is consistent with previous work in Legionella species suggesting that these proteins are under functional selection (67).
Key challenges in the detection of obligate intracellular pathogens are the difficulty in culturing the organisms and the lack of diagnostic targets that can detect these bacteria at low concentrations in often complex metagenomic samples. Here, we identified that Candidatus P. cheracis possesses a high copy transposase that had high sequence similarity to a symbiont of Solemya velum, the Atlantic awning clam. Future efforts to screen for the presence of Candidatus P. cheracis could be facilitated by PCR amplification of the transposase in aquatic samples. This would be akin to the diagnostic efforts of members of the Bordetella genus (68). Bordetella pertussis, the causative agent of whooping cough, is commonly identified through the PCR of high copy number insertion sequences (IS) with the primary target IS481. This would provide a potentially non-invasive, sensitive screening strategy for ongoing surveillance of the crayfish farms that would facilitate the prevention and absence of this pathogen in aquaculture environments. Additionally, if proven as a sensitive strategy for the detection of Candidatus P. cheracis, this PCR approach could also be employed to test different aquatic environments for the presence of this Coxiellaceae genus.
Comparative genomics of 65 Legionella species has reported an expansive repertoire of putative Dot/Icm effector proteins (69). While many species possess putative effectors with specific eukaryotic domains, such as GTPase, F-box, and SET domains, these motifs are encoded within different proteins that are not orthologous (69). Interestingly, only 8 Dot/Icm substrates are conserved among these 65 species of Legionella (69). Using two distinct in silico tools, we have developed a list of 238 putative Dot/Icm effectors of Candidatus P. cheracis. Unlike Legionella species, very few putative Dot/Icm effectors encoded by Candidatus P. cheracis possess similarity to eukaryotic domains, and a large proportion, ~28%, are novel. We observed 12 predicted effectors with similarity to known or putative C. burnetii Dot/Icm effectors, including CpeB and CvpE, which have been shown to make important contributions to the intracellular success of C. burnetii (48, 54, 59, 65). Further expanding the collection of sequenced Coxiellaceae genomes will facilitate greater comparison of the effector repertoire in this family, allowing us to determine whether some of these 12 common effectors act as “core” Coxiellaceae effectors that facilitate their characteristic interaction with eukaryotic cells. We were able to demonstrate Dot/Icm-dependent translocation of Candidatus P. cheracis CpeB (PcCpeB) using C. burnetii as a surrogate host. In the absence of methodology for the isolation and cultivation of Candidatus P. cheracis, the success of this approach provides an alternative strategy for developing our understanding of the host-pathogen interactions mediated by Candidatus P. cheracis.
The full genome sequence of Candidatus P. cheracis has highlighted potential screening or diagnostic targets and unveiled likely pathogenesis strategies. In addition, the genome sequence shows that Candidatus P. cheracis encodes for Mip, a peptidyl-prolyl cis-trans isomerase that has been shown to be essential for intracellular replication and pathogenesis of C. burnetii (70). Candidatus P. cheracis Mip shares 66% identity with C. burnetii Mip, and these proteins are conserved within the PPIase catalytic domain, which suggests that Mip inhibitors developed by Debowski et al. (70) may inhibit Candidatus P. cheracis Mip. Treatment with rationally designed small molecule inhibitors of Mip may represent a plausible intervention measure for future outbreaks of Candidatus P. cheracis in aquaculture facilities.
Here, we have further demonstrated the effective use of ONT sequencing to reconstruct the genome of a novel intracellular pathogen—a member of a family that is difficult to culture in standard laboratory conditions—directly from its host. The complete genome of a chromosome and two plasmids enabled the exploration of a previously unexplored pathogen that is important for Australian aquaculture. ONT was advantageous here for the capture of the two plasmids, elements that are often missed by short-read metagenomic binning approaches but can be critical for virulence (71, 72) and for dealing with the significant number of IS elements throughout this genome (73). The detection of a T4SS and cohort of novel effector proteins that likely facilitate the intracellular success of the pathogen provides the first insights into the host-pathogen interactions of this novel pathogen. ONT sequencing has shown promise in recent years for outbreak investigations due to its low cost, portability, minimal sample preparation, and rapid, real-time data output (74). If such an outbreak were to occur today, in addition to traditional histopathology, ONT metagenomics would enhance efforts to detect and characterize unknown emerging pathogens.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Dragičević P, Bielen A, Petrić I, Hudina S. 2021. Microbial pathogens of freshwater crayfish: a critical review and systematization of the existing data with directions for future research. J Fish Dis 44:221–247. doi:10.1111/jfd.1331433345337 · doi ↗ · pubmed ↗
- 2Ketterer PJ, Taylor DJ, Prior HC. 1992. Systemic rickettsia-like infection in farmed freshwater crayfish, Cherax quadricarinatus, p 173–179. In Shariff M, Subasinghe RP (ed), Diseases in Asian aquaculture. Asian Fisheries Society.
- 3Owens L, Muir P, Sutton D, Wingfield M. 1992. The pathology of microbial diseases in tropical Australian Crustacea, p 165–172. In Shariff M, Subasinghe RP (ed), Diseases in Asian aquaculture. Asian Fisheries Society.
- 4Tan CK, Owens L. 2000. Infectivity, transmission and 16S r RNA sequencing of a rickettsia, Coxiella cheraxi sp. nov., from the freshwater crayfish Cherax quadricarinatus. Dis Aquat Organ 41:115–122. doi:10.3354/dao 04111510918979 · doi ↗ · pubmed ↗
- 5Cooper A, Layton R, Owens L, Ketheesan N, Govan B. 2007. Evidence for the classification of a crayfish pathogen as a member of the genus Coxiella. Lett Appl Microbiol 45:558–563. doi:10.1111/j.1472-765X.2007.02240.x 17908227 · doi ↗ · pubmed ↗
- 6Ruth Elliman J, Owens L. 2020. Confirmation that candidatus Coxiella cheraxi from redclaw crayfish (Cherax quadricarinatus) is a close relative of Coxiella burnetii, the agent of Q-fever. Lett Appl Microbiol 71:320–326. doi:10.1111/lam.1333632491210 · doi ↗ · pubmed ↗
- 7Lührmann A, Newton HJ, Bonazzi M. 2017. Beginning to understand the role of the type IV secretion system effector proteins in Coxiella burnetii pathogenesis. Curr Top Microbiol Immunol 413:243–268. doi:10.1007/978-3-319-75241-9_1029536362 · doi ↗ · pubmed ↗
- 8Graells T, Ishak H, Larsson M, Guy L. 2018. The all-intracellular order legionellales is unexpectedly diverse, globally distributed and lowly abundant. FEMS Microbiol Ecol 94:fiy 185. doi:10.1093/femsec/fiy 18530973601 PMC 6167759 · doi ↗ · pubmed ↗