Space: reconciling multiple spatial domain identification algorithms via consensus clustering
Daoliang Zhang, Wenrui Li, Xinyi Sui, Na Yu, Shan Wang, Zhiping Liu, Xiaowo Wang, Zhiyuan Yuan, Rui Gao, Wei Zhang

TL;DR
This paper introduces Space, a method that combines multiple spatial domain identification algorithms to produce more consistent and reliable results in spatial transcriptomics.
Contribution
Space introduces a consensus clustering approach with novel loss functions to improve reliability and consistency in spatial domain identification.
Findings
Space effectively resolves inconsistencies among different clustering algorithms on spatial transcriptomics data.
The method achieves highly reliable clustering outputs across multiple public datasets.
Space provides flexible tools for downstream analysis like visualization and gene analysis.
Abstract
The rapid development of spatially resolved transcriptomics (SRT) technologies has provided unprecedented opportunities for characterizing and understanding tissue architecture. As this field continues to advance, various methods have been developed to computationally identify spatial domains within tissues. However, the performance of different algorithms on the same dataset is not always consistent. This inconsistency makes it difficult for researchers to select the most reliable results for downstream analysis. To address this challenge, we propose a domain identification method named Space. Space measures consistency between different methods to select reliable algorithms. It then constructs a consensus matrix to integrate the outputs from multiple algorithms. We introduce similarity loss, spatial loss, and low-rank loss in Space to enhance the accuracy and optimize computational…
Click any figure to enlarge with its caption.
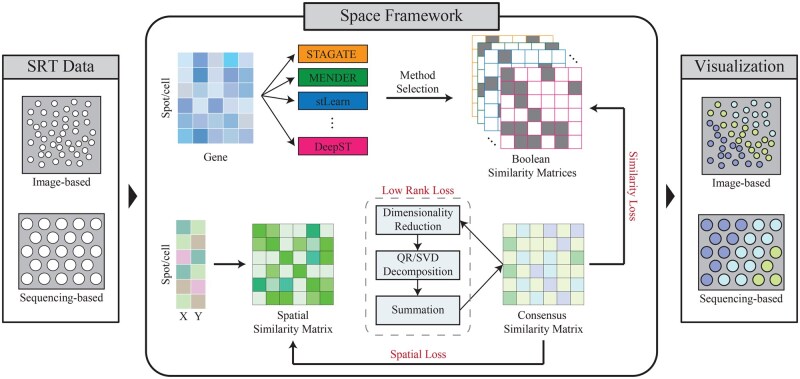 Figure 1
Figure 1- —National Natural Science Foundation of China10.13039/501100001809
- —Natural Science Foundation of Shandong Province10.13039/501100007129
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGene expression and cancer classification · Single-cell and spatial transcriptomics · Molecular Biology Techniques and Applications
1 Introduction
Characterizing tissue regions from a spatial perspective is essential for deciphering tissue development and understanding biological mechanisms. The continuous advancement of spatially resolved transcriptomics (SRT) technologies has provided a robust data foundation, enabling the identification of structural patterns within tissue. In recent years, various computational methods have been proposed for identifying spatial domains in SRT data. For instance, STAGATE (Dong and Zhang 2022) and MENDER (Yuan 2024) integrate gene expression information with spatial locations to decipher tissue structure. Methods like stLearn (Pham et al. 2023) and DeepST (Xu et al. 2022) perform multi-modal clustering analysis by combining gene expression profiles, spatial locations, and morphological features.
There is no doubt that each of these methods has its own unique strategies and characteristics. However, the performance of different algorithms on the same dataset is not always consistent. To address this problem, we propose a spatial domain identification framework called Space. In Space, we integrate 10 widely-used spatial domain identification methods. Additionally, we employ consensus clustering to combine the outputs of the reliable methods, achieving optimal domain identification results. To address the challenge of determining the number of spatial domains in unlabeled datasets, we propose a strategy for estimating the number of spatial domains. Space also offers a variety of evaluation metrics and user-friendly interfaces for downstream analysis.
2 Materials and methods
The flowchart of Space is shown in Fig. 1. First, we select ten methods based on their performance, popularity, and methodological diversity, and applied them to the SRT datasets. Next, we identify algorithms with consistent results and convert these results into Boolean similarity matrices which represent whether different cells are classified into the same spatial domain. Simultaneously, considering that spatially adjacent cells typically share functional similarities, we introduce a spatial similarity matrix. Next, Space constructs a consensus similarity matrix and minimizes the similarity loss and spatial loss with the two aforementioned matrices respectively to obtain the consensus result. To reduce noise and improve model stability, Space also incorporates a low-rank loss through nuclear norm constraint. Finally, spectral clustering is adopted to identify spatial domains. For more detailed information, please see Supplementary Materials.
The workflow of Space.
We conducted comprehensive tests on SRT datasets from different platforms. The experimental results demonstrate that Space effectively combines the strengths of different algorithms, obtaining precise and robust results (Supplementary Figs S1–S4).
For user convenience, Space offers a simple deployment process. The entire functionality can be executed with a few Python commands. Additionally, Space supports input and output compatibility with current mainstream analysis software, such as SCANPY (Wolf et al. 2018), enabling seamless downstream analyses for users from different fields.
3 Conclusion
Here, we propose Space as a highly integrated spatial domain identification toolkit. Users can run it without the need for complex parameter tuning. This toolkit not only helps researchers quickly decipher tissue structures but also facilitates downstream analysis of SRT data in future studies.
Supplementary Material
vbaf084_Supplementary_Data
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Dong K , Zhang S. Deciphering spatial domains from spatially resolved transcriptomics with an adaptive graph attention auto-encoder. Nat Commun 2022;13:1739.35365632 10.1038/s 41467-022-29439-6PMC 8976049 · doi ↗ · pubmed ↗
- 2Pham D , Tan X, Balderson B et al Robust mapping of spatiotemporal trajectories and cell–cell interactions in healthy and diseased tissues. Nat Commun 2023;14:7739.38007580 10.1038/s 41467-023-43120-6PMC 10676408 · doi ↗ · pubmed ↗
- 3Wolf FA , Angerer P, Theis FJ. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol 2018;19:15.29409532 10.1186/s 13059-017-1382-0PMC 5802054 · doi ↗ · pubmed ↗
- 4Xu C , Jin X, Wei S et al Deep ST: identifying spatial domains in spatial transcriptomics by deep learning. Nucleic Acids Res 2022;50:e 131.36250636 10.1093/nar/gkac 901PMC 9825193 · doi ↗ · pubmed ↗
- 5Yuan Z. MENDER: fast and scalable tissue structure identification in spatial omics data. Nat Commun 2024;15:207.38182575 10.1038/s 41467-023-44367-9PMC 10770058 · doi ↗ · pubmed ↗