Phylogenetic taxonomy of the Zambian Anopheles coustani group using a mitogenomics approach
Soha Usmani, Mary E. Gebhardt, Limonty Simubali, Kochelani Saili, Westone Hamwata, Hunter Chilusu, Mbanga Muleba, Conor J. McMeniman, Anne C. Martin, William J. Moss, Douglas E. Norris, Reneé L.M.N. Ali

TL;DR
This study uses complete mitochondrial genomes to better classify and understand the evolutionary history of Anopheles coustani mosquitoes in Zambia.
Contribution
The study introduces mitogenomics as a powerful tool for resolving taxonomic confusion in the Anopheles coustani group.
Findings
Seventeen new complete mitogenomes were generated, comparable in structure to reference An. coustani mitogenomes.
Bayesian phylogenetic analysis identified six distinct clades, including a previously unknown species.
Divergence times suggest the An. coustani group split from the An. gambiae complex around 110 million years ago.
Abstract
Mosquito species belonging to the Anopheles coustani group have been implicated in driving residual malaria transmission in sub-Saharan Africa and are regarded as an established primary vector in Madagascar. The morphological identification of mosquitoes in this group is challenging due to cryptic features and their molecular confirmation is difficult due to a paucity of reference sequence data representing all members of the group. Conventional molecular barcoding with the cytochrome oxidase I (COI) gene and the internal transcribed spacer 2 (ITS2) region targets is limited in their discrimination and conclusive identification of members of species complexes. In contrast, complete mitochondrial genomes (mitogenomes) have demonstrated much improved power over barcodes to be useful in rectifying taxonomic discrepancies in Culicidae. We utilized a genome skimming approach via shallow…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
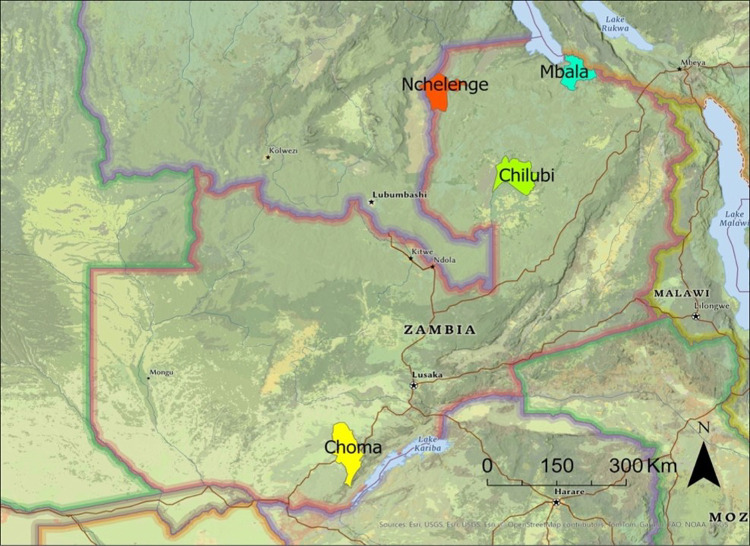 Figure 1
Figure 1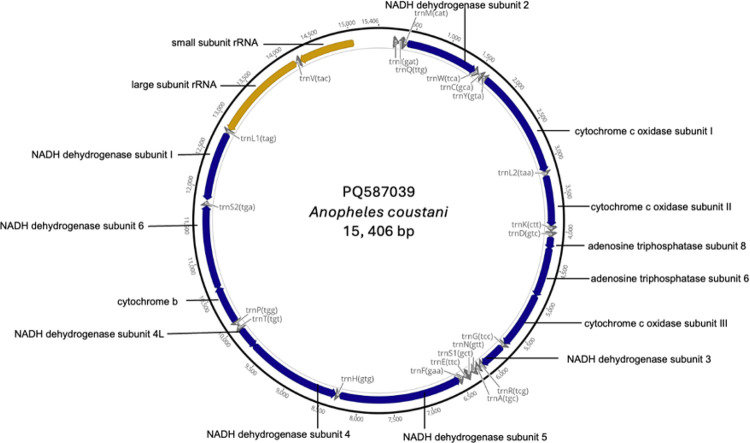 Figure 2
Figure 2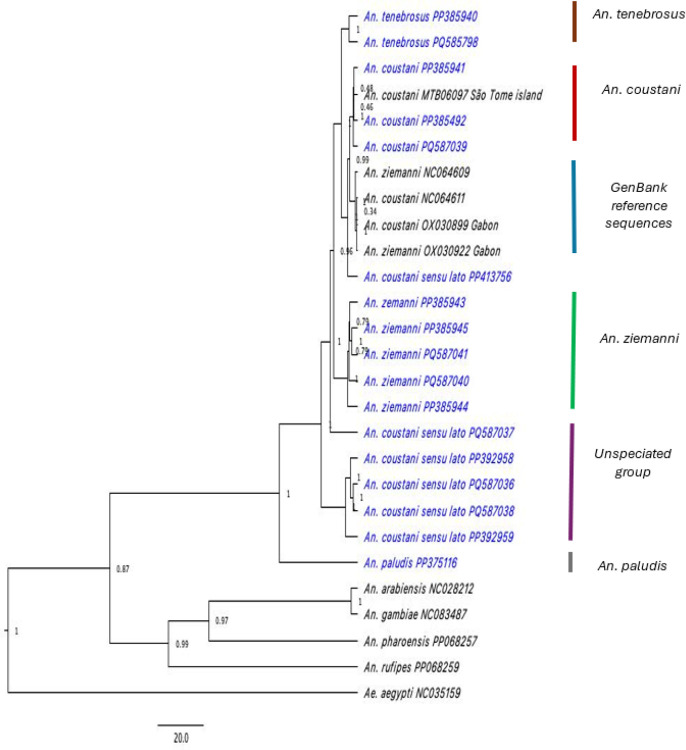 Figure 3
Figure 3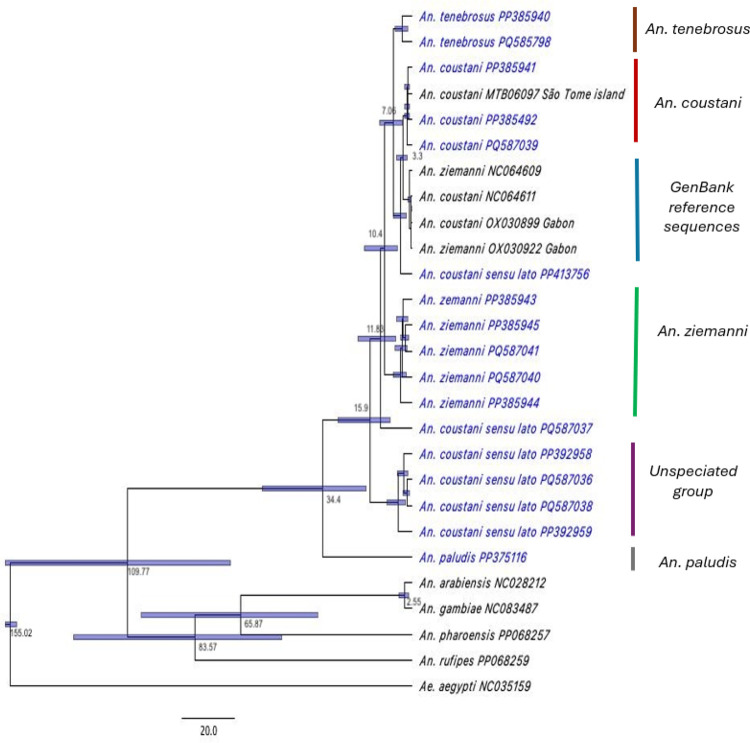 Figure 4
Figure 4Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMosquito-borne diseases and control · Malaria Research and Control · Insect symbiosis and bacterial influences
Background
Vector control methods like indoor residual spraying (IRS) and long-lasting insecticidal nets (LLINs) have been instrumental in progress toward malaria elimination [1, 2]. Primary, well-studied vectors like Anopheles gambiae and An. funestus, which typically engage in endophagic and endophilic behaviors by seeking human hosts indoors, are the focus of these key intervention measures [1, 2]. However, selection pressure driven by the broad deployment of IRS and LLINs have either reduced these populations, driven insecticide resistance, yielded shifts in vector species composition and/or resulted in changes in biting and resting behaviors [2–7]. Shifts to outdoor biting or having a high plasticity in this behavior, and the existence of other exophagic malaria vectors have been identified as a significant barriers to malaria control and elimination [3, 8, 9]. Though frequently collected, exophagic anopheline mosquitoes such as members of the An. coustani group [10–12], An. squamosus, and An. rufipes [14] are understudied despite contributing to malaria transmission in sub-Sahran Africa.
The Anopheles coustani group is widely distributed throughout sub-Saharan Africa and the Middle East, with members typically exhibiting zoophilic and outdoor foraging behaviors [11]. Within the group, morphologically similar species including An. coustani, An. zeimanni, An. paludis, and An. tenebrosus, have demonstrated opportunistic foraging towards anthropophilic and endophilic feeding [10, 15]. Little is known about the basic biology, ecology and behaviors of most of these species. This knowledge gap is particularly noteworty given members of the group have been implicated as established vectors with a key role in sustaining residual malaria transmission in Kenya, Madagascar, Ethiopia, Cameroon, Mozambique and Zambia [10, 15–20]. Members of this group present an imminent threat to malaria elimination efforts due to inherent plasticity in their foraging behaviors, which enable them to evade many of the existing vector control strategies that target endophagic and endophilic mosquitoes [3, 21–23].
Morphological and molecular techniques have proved to be challenging for identification of species in this group due to cryptic features, damaged specimens which obscures key morphological attributes [23–25], and the paucity of reference molecular data for comparison in genomic repositories [26]. Additionally, the well-established cytochrome oxidase I gene (COI) and the internal transcribed spacer 2 (ITS2) molecular barcodes commonly used for species confirmation have limited power in delineating phylogenetic disparities in cryptic species groups [23, 27, 28]. Though limited in number, published genetic and molecular studies have highlighted cryptic members within the An. coustani group [29–31]. Early studies using chromosomal inversion analyses identified An. coustani and An. crypticus as separate species [29, 30]. Genetic diversity analyses in Zambia and the Democratic Republic of the Congo also reported two distinct phylogenetic groups of An. coustani populations [31] in 2020, and definitive species identification remained unverified based on conventional barcoding methods in Mozambique in 2024 for An. tenebrosus and An. Zeimanni [18].
Mitochondrial genomes (mitogenomes) are circular, double stranded DNA molecules with high copy numbers, low incidence of recombination, absence of introns, and maternal inheritance [32–34]. These characteristics facilitate utility for inferring phylogenies, addressing species identification, and evolutionary studies in a range of organisms including metazoans [35–37]. The mitogenome encodes for 13 protein coding genes (PCGs), 22 transfer RNA (tRNA), 2 ribosomal RNA (rRNA) and a non-coding control region [38]. Developments in computational and sequencing technologies enable more datasets to include chromosomal and mitochondrial reference genomes for mosquito species, where both data are available [32, 36, 39, 40]. However, sequencing efforts to date have been biased toward well-studied and defined species groups such as An. gambiae [41–43] and An. funestus [44, 45].
At the present time, there are five mitochondrial and two chromosomal genomes collectively available in the GenBank databse for An. coustani sensu stricto and An. ziemanni[46–49]. Generating additional reference mitogenomes for members of the An. coustani group would prove beneficial for phylogenetic analyses and these data can inform taxonomic classification, mosquito diversity, and evolutionary history in relation to malaria transmission of this understudied group [50, 51]. Although full genomes would be ideal for these tasks, mitochondrial genomes can be sequenced and assembled quickly and inexpensively compared to full nuclear genome sequencing and annotation.
Given that accurate species identification is crucial for vector incrimination and the development and evaluation of vector control strategies, the taxonomic resolution of species in the An. coustani group is essential for malaria control efforts [23]. Additionally, it is not plausible to generate significant inferences regarding population and evolutionary histories or actual taxonomic species boundaries based on currently available evidence. This study aims to contribute complete reference mitochondrial genomes for members of the An. coustani group in Zambia and delineate the phylogenetic taxonomy for this epidemiologically important mosquito complex.
Methods
Mosquito collection and morphological identification.
Outdoor mosquito collections were carried out in Zambia as part of the Southern and Central Africa International Centers of Excellence for Malaria Research (ICEMR). Specimen collections were performed in 2023–2024 using standard Centers for Disease Control and Prevention (CDC) miniature light traps in Choma and Nchelenge Districts (Fig. 1). Larvae were collected in the Chilubi and Mbala Districts and were reared to adults at the Tropical Diseases Research Centre (TDRC), Ndola, Zambia. Mosquitoes were sorted and identified using a morphological key [52] by members of the ICEMR team. Specimens morphologically identified as An. coustani, An. ziemanni, An. tenebrosus, and An. paludis were stored in tubes containing silica gel and shipped to the Johns Hopkins Bloomberg School of Public Health (Maryland, USA) for molecular analysis. The specimens with intact morphological characteristics that allowed clear identification as An. coustani, An. tenebrosus, An. paludis and An. ziemanni, were molecularly confirmed and selected for sequencing and downstream analysis. Specimens that could not be further keyed to species type due to damage or cryptic features were labelled as An. coustani sensu lato (s.l.).
DNA extraction, sequencing, mitogenome assembly and annotation.
Single mosquito specimens were homogenized in a mixture containing 98 μL of PK buffer (Applied Biosystems, Waltham, MA) and 2 μL of proteinase K (Applied Biosystems, Waltham, MA) followed by an incubation at 56°C for 2.5 hours [53]. After incubation, DNA was extracted using the Qiagen DNeasy Blood and Tissue kit (Qiagen, Hilden, Germany) as per the manufacturer’s instructions. Using the Qubit dsDNA assay kit (Thermo Fisher Scientific, Waltham, MA) the extracted DNA was quantified and stored at −20°C. Extracted DNA was shipped to SeqCenter (Pittsburgh, USA) for library construction and Illumina sequencing. Libraries were 150 bp paired end sequenced to a depth of 13.3 million reads.
Using NOVOPlasty [54] (RRID:SCR_017335) version 4.3.5, the mitochondrial genomes were assembled with k-mer set at 39 and reference mitogenomes (MT_806097, NC_064609, NC_064611) as seed sequences. The generated contigs were automatically annotated using the MITOchondrial genome annotation (MITOS) [55] galaxy tool under the invertebrate genetic code with default settings. Using reference An. coustani mitochondrial genomes as guides, start and stop codon positions were manually modified in Geneious Prime (RRID:SCR_010519) version 2023.2.1 (Biomatters, Auckland, Australia). Resulting sequences and their corresponding annotations were uploaded to the GenBank database.
Phylogenetic analysis and divergence time estimation
The protein coding genes of the mitogenomes constructed in this study and those from An. coustani (MT_806097, NC_064611, OX_030899), An. ziemanni (NC_064609, OX_030922), An. gambiae (NC_083487), An. arabiensis (NC_028212), An. pharoensis (PP_068257), An. rufipes (PP_068269) and Ae. aegypti (NC_035159) reference sequences were imported from the GenBank repository, aligned, and exported in nexus format using the MAFFT amino acid alignment mode in Geneious Prime (RRID:SCR_010519) version 2023.2.1 (Biomatters, Auckland, Australia). Using jModelTest (v2.1.10) software [56] with default settings. The best fit base pair substitution model for the aligned sequence matrix was identified based on the Bayesian information criterion (BIC) and the Akaike information criterion (AIC). Bayesian inference analysis and node age calculations were performed in Bayesian Evolutionary Analysis by Sampling Trees (BEAST) version 2.7.6 [57] using the GTR + G + I substitution model with three independent runs as described [58]. An application of 20% burn-in rate was implemented for tree building purposes and FigTree v.1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/) was used to visualize trees. Molecular dating time estimations were inferred alongside the previously mentioned parameters using Aedes-Anopheles divergence time as the calibration point. The Aedes-Anopheles divergence was set as a prior with normal distribution around 154.7 million years ago (MYA) [59]. Pairwise genetic distances between representative groups were computed in the MEGA X 10.0.5 software [60] using the exported MAFTT amino acid alignment from Geneious Prime.
Results
Mitochondrial genome characteristics
Review of collections from 2023–2024 provided 81 putative An. coustani group specimens. From these, 17 specimens passed morphological and molecular confirmation, and were sequenced and annotated. The 17 novel mitogenomes produced in this study were arranged similarly to the reference An. coustani and An. ziemanni mitochondrial genomes available in the GenBank database, with lengths ranging from 15,404 bp (An. tenebrosus) to 15,425 bp (An. paludis) and an average AT content of 78.3% (Table S1). The An. coustani group mitogenomes comprised of 13 PCGs, 22 transfer RNAs (tRNAs) and 2 ribosomal RNAs (rRNAs) as shown in Fig. 2.
Phylogenetic and Divergence time analysis
The aligned and concatenated protein coding sequences from the 25 mitogenomes (24 Anopheles and 1 Aedes mosquito species as an outgroup) resulted in a matrix of 11,023 bp, which was included in the Bayesian analyses for the phylogenetic tree construction and molecular dating. Bayesian inferences resulted in well supported phylogenies with posterior probabilities close to or at one for the mitogenomes generated in this study. Six main clades were identified. Five clades represent four taxa (An. tenebrosus, An. coustani, An. ziemanni, An. paludis) from the An. coustani group and an ‘unspeciated’ group comprised of specimens morphologically identified as An. coustani s.l. (Fig. 3). The sixth clade is comprised of the GenBank reference sequences labeled as An. coustani and An. ziemanni as identified in GenBank.
The most recent common ancestor (MRCA) of all Anopheles was dated at 109.77 MYA (Fig. 4) with a 95% confidence interval spanning from 68.4 to 157.02 MYA (Table 1), using the Anopheles-Aedes divergence period set at 154.7 MYA [59]. The MRCA for An. coustani s.l. and An. ziemanni within the An. coustani group dates to 10.4 MYA, with a credibility interval that spans from 0.7 to 14.3 MYA. This MRCA is more recent than those determined for the unspeciated group and An. paludis from other members of the An. coustani group, estimated at 15.9 and 34.4 MYA respectively (Fig. 4 and Table 1). The pairwise genetic distance matrix calculations (Table S2) between representatives of each group/clade ranged from 0.0008–0.0217, except for An. paludis which resulted in a much wider genetic distance.
Discussion
This study generated 17 new full-length mitochondrial genomes for members of the An. coustani group from Zambia that improve the resolution of within-group species taxonomy and provide insight into the species group’s complexity. Bayesian analyses using the concatenated PCGs from the mitogenomes generated in this study supported phylogenies and separated the specimens into distinct taxonomic groups including An. coustani s.s., An. tenebrosus, An. paludis and An. ziemanni. These new phylogenies have better taxonomic resolution and stronger branch support when compared to earlier studies in Zambia using the COI and ITS2 molecular barcodes [31, 61]. Those studies separated An. coustani s.l. specimens into two general groups, An. coustani clade 1 or 2 [31, 61], or undefined Anopheles species groups [61]. Furthermore, a subset of the An. coustani s.s. specimens in this study formed a separate clade from the GenBank reference genome sequences identified as An. coustani and An. ziemanni, an indication of additional complexity within the An. coustani species group or perhaps, morphological misidentification prior to sequencing.
This study highlights the significance of anopheline morphological data and molecular verification for identifying both known and unknown anopheline species, especially those implicated as malaria vectors. Though previous studies have shed light on mosquitoes in the An. coustani group and their association with malaria transmission [20, 21, 23, 31], there remains a paucity of sequence data corresponding to well-curated specimens which can be used to accurately speciate members of this group. As a result, the majority of available COI and ITS2 sequences are categorized as ‘An. coustani s.l.’, rather than to specific species within the group [18, 24, 61].
Despite the increased taxonomic power the data in this study provided, there were some limitations to identification of all specimens. In the absence of voucher specimens available for sequencing or genomic data for other members of the group such as An. caliginosus, An. crypticus, An. namibiensis and An. symesi [11] our study faced challenges in determining the phylogenetic placement and species identification for one clade of specimens, which we designated as An. coustani s.l. These mosquito specimens were collected primarily in Nchelenge District on the border with the Democratic Republic of the Congo (DRC) where An. caliginosus has been reported [11, 62], suggesting this species or perhaps other members of the An. coustani group may be more widely distributed in Zambia. Another caveat is the indistinguishable morphological features of adult female An. crypticus and An. coustani s.s. mosquitoes [11, 30]. It is possible that the An. coustani s.s. specimens sequenced in this study, or alternatively the GenBank references, represent An. crypticus. This was implied by a study that identified ‘An. coustani clade 2’ as putative An. crypticus [61]. Furthermore, pairwise distance estimates between representatives from these two groups suggest the potential prescence of An. crypticus circulating in Zambia. However, with the lack of reference specimens and the documented species range limited to South Africa [11, 30], it is problematic to verify the presence of this species or correlate molecular and cytogenetic data to morphological identifications across different species and studies.
Genetic distance matrices may provide definition of species boundaries [63], and the calculations derived from this study reinforce the complexity of relatedness among species such as An. coustani and An. ziemanni, further implying that cryptic speciation may be due to behavioral and ecological preferences [64]. Although studies for African anophelines have been biased towards well-recognized vectors such as An. funestus and An. gambiae [43, 45, 65], divergence estimations and phylogenies are also reported to be unresolved due to complexities such as introgression [25, 58, 66]. Our molecular divergence calculations suggest the An. coustani group diverged from the An. gambiae species complex ~110 MYA. This is consistent with inferences made by previous studies which reported the last common ancestor of Anopheles ~100 MYA [67] and the African distribution of the Anopheles subgenus ~113 MYA [68]. Molecular dating based on this phylogenetic analysis shows An. paludis splitting ~ 34 MYA from closely related species group members. This divergence time is older than that estimated between the other clades and like that for An. gambiae and An. funestus, suggests that reproductive or opportunistic behavioral adaptions may have occurred to explain why some species group members may be more involved in the transsion of Plasmodium falciparum.
Conclusions
This is the first publication using a genome skimming strategy to generate 17 mitochondrial genomes for representatives of the An. coustani group. We were able to estimate divergence times for members of the group for which there is data and this study emphasizes the importance of actively pursuing accurately identified morphological voucher specimens for molecular characterization collected from other African regions. This is required for the clear delineation of species boundaries as well as for the taxonomic rectification among An. coustani members which have been shown to be closely related in this study. These findings also highlight the need for study of the basic biology of this group, inlcuding reproductive compatibility between members of the group which may resolve some of the taxonomic mysteries and most critically, their biological capacity to vector human pathogens is largely unknown. With changes in land use, climate and the decrease or shifts in primary malaria vector populations, research should focus on the ecological and behavioral characteristics of species in this and similarly understudied anopheline groups, as their importance in malaria transmission becomes more prominent.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Pryce J, Medley N, Choi L. Indoor residual spraying for preventing malaria in communities using insecticide-treated nets. Cochrane Database Syst Reviews. 2022;17(1):1.10.1002/14651858.CD 012688.pub 3PMC 876303335038163 · doi ↗ · pubmed ↗
- 2Sherrard-Smith E, Ngufor C, Sanou A, Guelbeogo MW, N’Guessan R, Elobolobo E, Inferring the epidemiological benefit of indoor vector control interventions against malaria from mosquito data. Nat Commun. 2022;13:3862.35790746 10.1038/s 41467-022-30700-1PMC 9256631 · doi ↗ · pubmed ↗
- 3Sougoufara S, Ottih EC, Tripet F. The need for new vector control approaches targeting outdoor biting anopheline malaria vector communities. Parasites Vectors. 2020;13:295.32522290 10.1186/s 13071-020-04170-7PMC 7285743 · doi ↗ · pubmed ↗
- 4Musiime AK, Smith DL, Kilama M, Rek J, Arinaitwe E, Nankabirwa JI, Impact of vector control interventions on malaria transmission intensity, outdoor vector biting rates and Anopheles mosquito species composition in Tororo, Uganda. Malar J. 2019;18:445.31881898 10.1186/s 12936-019-3076-4PMC 6935116 · doi ↗ · pubmed ↗
- 5Sherrard-Smith E, Skarp JE, Beale AD, Fornadel C, Norris LC, Moore SJ Mosquito feeding behavior and how it influences residual malaria transmission across Africa. Proceedings of the National Academy of Sciences. 2019;116:15086–95.10.1073/pnas.1820646116 PMC 666078831285346 · doi ↗ · pubmed ↗
- 6Kreppel KS, Viana M, Main BJ, Johnson PCD, Govella NJ, Lee Y, Emergence of behavioural avoidance strategies of malaria vectors in areas of high LLIN coverage in Tanzania. Sci Rep. 2020;10:14527.32883976 10.1038/s 41598-020-71187-4PMC 7471940 · doi ↗ · pubmed ↗
- 7Sanou A, Nelli L, Guelbeogo WM, Cisse F, Tapsoba M, Ouedraogo P, Insecticide resistance and behavioural adaptation as a response to long-lasting insecticidal net deployment in malaria vectors in the Cascades region of Burkina Faso. Sci Rep. 2021;11:17569.34475470 10.1038/s 41598-021-96759-w PMC 8413378 · doi ↗ · pubmed ↗
- 8Reddy MR, Overgaard HJ, Abaga S, Reddy VP Caccone A, Kiszewski AE, Outdoor host seeking behaviour of Anopheles gambiae mosquitoes following initiation of malaria vector control on Bioko Island, Equatorial Guinea. Malar J. 2011;10:184.21736750 10.1186/1475-2875-10-184PMC 3146901 · doi ↗ · pubmed ↗