Neurofibromatosis-Noonan syndrome: a prospective monocentric study of 26 patients and literature review
Didier Bessis, Dominique Vidaud, Pierre Meyer, Laurence Pacot, de La Villeon G, Adeline Alice Bonnard, Yline Capri, Christine Coubes, Fanchon Herman, Didier Lacombe, Nicolas Molinari, Laura Poujade, Agathe Roubertie, Julien Van Gils, Alain Verloes, David Geneviève, Hélène Cavé

TL;DR
This study identifies key clinical features of Neurofibromatosis-Noonan syndrome and highlights the importance of cardiovascular screening and facial recognition tools in diagnosis.
Contribution
The study provides a detailed clinical characterization of NF-NS and validates the use of Face2Gene® for diagnostic agreement.
Findings
NF-NS is marked by Noonan-like facial features and a high frequency of pectus excavatum.
Face2Gene® showed high diagnostic concordance with clinicians for facial phenotype assessment.
Congenital heart malformations, particularly pulmonic stenosis, are common in NF-NS patients.
Abstract
Data on clinical manifestations of neurofibromatosis-Noonan syndrome (NF-NS) remain heterogeneous, with limited validated descriptions. This study aims to better define the clinical and molecular features of NF-NS and compare them with existing literature. Secondary objectives include evaluating inter-rater diagnostic agreement among experienced clinicians and assessing the utility of deep-learning algorithms (Face2Gene® [F2G]). Additionally, we assess the prevalence of congenital heart malformations (CHM) in NF-NS compared to ‘classic’ neurofibromatosis type 1 (NF1). A 9-year, prospective, monocentric study was conducted, involving patients with NF1 pathogenic variants (PVs) and Noonan syndrome-like facial phenotype (NSLFP). Twenty-six patients were enrolled. NSLFP was categorized as ‘suggestive’ in 69% of cases and ‘typical’ in 31%. The presence of at least two facial abnormalities…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
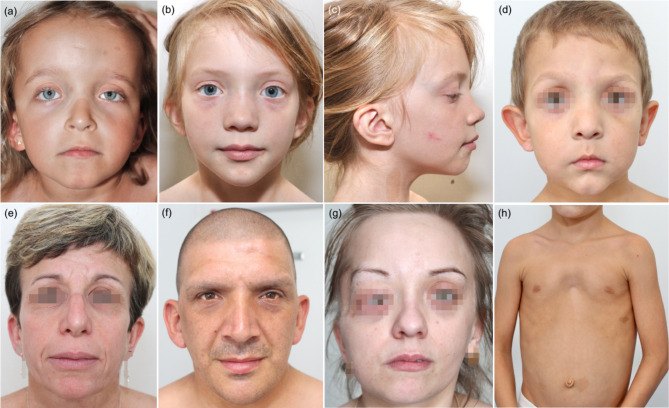 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsNeurofibromatosis and Schwannoma Cases · Protein Tyrosine Phosphatases
Introduction
Neurofibromatosis-Noonan syndrome (NF-NS; OMIM # 601321) is a rare autosomal-dominant disorder, first described by Allanson et al. in 1985 [1]. It presents with clinical features overlapping those of neurofibromatosis type 1 (NF1; OMIM # 162200) and Noonan syndrome (NS; OMIM # 163950). Approximately 320 cases identified as NF-NS have been reported (Table S1) [2–52]. Molecular studies of the NF1 gene in NF-NS, initiated in 2005, has established that NF-NS is consistently a phenotypic variant of NF1, characterized by a high prevalence of missense or in-frame deletions of pathogenic variants (PVs) in the NF1 gene.
However, the literature on NF-NS remains heterogeneous, mainly due to the lack of validated diagnostic criteria. Consequently, diagnosis has often been based on case reports or studies conducted under non-comparable conditions. Diagnosis were made based on: (i) the presence of ‘Noonan syndrome-like’ facial phenotype (NSLFP), often without detailed clinical descriptions; (ii) other NS diagnostic criteria, such as short stature, pectus deformity, or pulmonic valve stenosis - features also commonly reported in ‘classic’ NF1; or (iii) in nearly half of all cases, large NF1 cohort studies with genotype-phenotype correlations linked to recurrent PVs, including p.(Met992del) in-frame deletion [23], p.(Met1149) [24], p.(Arg1809) [31, 38], p. (Arg1038) [22, 45], p.(Arg1276) [24], p.(Lys1423) [24], and codons 844–848 missense PVs [22]. Moreover, cases with PVs such as p.(Arg1276) [24], p.(Lys1423) [24], and p.(Arg1809) [31] missense PVs appear to be associated with a higher prevalence of congenital heart malformations (CHM), including pulmonic valvular stenosis (PVS), compared to “classic” NF1.
Given these diagnostic challenges, it is important to clarify whether specific clinical features, such as CHM, are more prevalent in NF1 patients with NSLFP compared to ‘classic’ NF1. This also raises the question of whether systematic cardiovascular evaluations, including follow-up by a cardiologist with echocardiography, should be recommended in NF1 with NSLFP, similar to guidelines for other RASopathies such as NS [53], cardiofaciocutaneous syndrome [54], and Costello syndrome [55].
In this French monocentric and multidisciplinary prospective study, our primary objective was to better define the clinical manifestations of NF-NS by studying a cohort of children and adults with molecularly confirmed NF1 PVs. Our secondary objectives were twofold: (i) to evaluate inter-rater agreement among clinicians experienced in diagnosing NF-NS and assess the effectiveness of phenotypic evaluation; and (ii) to determine whether there is an increased risk of CHM in NF-NS patients, regardless of the type of NF1 PV, compared to those with ‘classic’ NF1.
Patients and methods
We prospectively enrolled children and adults with suspected NF-NS, evaluated at the Reference Center for Rare Skin Diseases and departments of medical genetics and pediatric neurology at CHU Montpellier, Montpellier, France, from March 2013 to December 2022.
This study was approved by the Clinical Research Department of the University Hospital (DB, MW) and relevant ethics committees. Informed consent was obtained from all participants or their legal guardians.
Inclusion and evaluation criteria
Patients were included if they had a clinically confirmed diagnosis of NF1, based on NIH diagnostic criteria (1988 and revised in 2021) [56, 57], and a confirmed NF1 PV, along with NSLFP. Each patient underwent a thorough clinical assessment, including family history, physical exams, and evaluations of the cutaneous, neurological, ophthalmological, skeletal, and cardiac systems.
Facial phenotype analysis
NSLFP was assessed by a dermatologist and geneticists (DB, DG and MW), considering the following NSLFP features: coarse facial features, flat occiput/brachycephaly, facial asymmetry, prominent and high forehead, frontal bossing, ptosis, hypertelorism (interpupillary distance > 2 standard deviations), midface hypoplasia, triangular face, downslanted palpebral fissures, eversion of the lateral eyelid, thickened eyelids, epicanthal folds, low-set posteriorly angulated ears, thickened upper helix, high and broad nasal bridge, depressed flat nasal root, bulbous and upturned nasal tip, hooked nose, wide and prominent philtrum, wide peaks to vermillion border of the upper lip (cupid’s bow appearance), micrognathia, and a small, pointed chin.
Standardized 2D facial images, including both front-facing and profile views, were taken during clinical visits using standard digital photography. To ensure a natural facial gesture, images were acquired in an upright position with a neutral facial expression. All photographic images were reviewed separately by a team of geneticists (MW, DG, DL, JVG, AV, YC) and dermatologist (DB). NSLFP was rated according to the following classification: typical (scored 2), suggestive (scored 1), and low-suggestive (scored 0).
Frontal images were analyzed using the Face2Gene^®^ (F2G) tool (FDNA Inc., Boston MA, USA, v.19.1.7) without any additional molecular or clinical information provided [58]. F2G is a clinical decision support tool that leverages machine learning to assist in the diagnosis of genetic syndromes. By analyzing facial photographs, the software compares the patient’s facial features to known genetic syndromes and generates a differential diagnosis listing the top 30 syndrome matches. For each syndrome, the software evaluates the images by creating a heat-map based on the Gestalt score confidence, categorizing the results as “high” (considered typical, scored 2), “medium” (considered suggestive, scored 1), or “low” (considered low-suggestive, scored 0).
Genetic screening
Genetic screening of genes known to be involved in RASopathies (i.e. PTPN11, SOS1, SOS2, SHOC2, CBL, HRAS, NRAS, KRAS, RIT1, RRAS, RRAS2, BRAF, RAF1, MAP2K1, MAP2K2, SPRED1, SPRED2, NF1, PPP1CB, and LZTR1) [59] was performed by next-generation sequencing (NGS) on genomic DNA obtained from peripheral leukocytes. Briefly, NGS was performed using capture-based target enrichment (Custom SureSelect XTHS2, Agilent) and sequencing on a NextSeq500^®^ (High Output Kit v2, 2150 bp) or NextSeq2000^®^ (Flow Cell P2, 2150 bp) (Illumina). Bioinformatic alignment was performed using Pipeline Local Run Manager v.2.4.0 (Illumina). Read alignment and variant calling was performed using VarScan v.2.3.5, with the UCSC GRCh37/hg19 genome assembly version as reference. Variant classification was performed using Alissa Interpret^®^ (Agilent Technologies). The average sequencing depth was 100x. The pathogenicity of amino acid variants was interpreted according to international expert consensus [60, 61], taking into consideration the Human Gene Mutation Database (HGMD), Leiden Open Variation Database (LOVD3.0) and ClinVar information.
NF1 variants were named according to the National Center for Biotechnology Information (NCBI) reference transcript sequence with the following GenBank accession number NF1 (NC_000017.10). Previous reports of single nucleotide variants were checked by consulting the Ensembl genome browser (http://www.ensembl.genome.org).
Statistical analysis
Categorical variables were reported with the number of observations (N) and the frequency of each modality (%). Group comparisons were made using the Chi-squared test or Fisher’s exact test, as appropriate. P-values were adjusted using the false discovery rate method.
Concordance analysis including both inter-rater between one dermatologist (DB) and six geneticists (AV, DG, DL, MW, JVG, YC) and the clinicians’ panel average rating versus F2G analysis was performed using Gwet’s AC coefficient. The interpretation of Gwet’s AC coefficient was as follows: < 0: poor agreement; 0.01–0.20: slight agreement; 0.21–0.40: fair agreement; 0.41–0.60: moderate agreement; 0.61–0.80: substantial agreement; 0.81–1.00: almost perfect agreement. The Gwet’s AC coefficient is presented with its 95% confidence interval (CI), and a summary of the various Gwet’s AC coefficients is displayed using a Forest Plot. All statistical tests were two-sided, and P-values ≤ 0.05 were considered statistically significant. All statistical analyses were performed using R software, version 4.3.1.
Results
From March 2013 to December 2022, 26 patients diagnosed with NF-NS were recruited, representing 4.7% of a cohort of 512 NF1 (NIH criteria, revised in 2021). The characteristics of these patients are summarized in Table 1 (details in Table S2). All patients were Caucasian and predominantly male (73%), with a median age of 10 years (range 1–45).
Table 1. Baseline characteristics and frequency of clinical manifestations end pathogenic variants NF1 in our series in comparison with the data from the literature on neurofibromatosis type 1-Noonan syndrome and neurofibromatosis type 1Neurofibromatosis type 1-Noonan syndromeOur studyNeurofibromatosis type 1-Noonan syndrome literature reviewNeurofibromatosis type 1 literature reviewOur study vs. neurofibromatosis type 1-Noonan syndrome literature review (Corrected P-value)Our study vs. neurofibromatosis type 1 literature review(Corrected P-value)Neurofibromatosis type 1-Noonan syndrome literature review vs. neurofibromatosis type 1 literature review(Corrected P-value)Baseline characteristicsNumber of patients26321ND---Sex ratio1.1 (19 M/17 F)1.1 (159 M/142F)ND1--Facial Noonan phenotype100% (26/26)100% (242/242)3.1% (93/2978)1 < 0.001
< 0.001 Down-slanting palpebral fissures53.8% (14/26)45.9% (111/242)ND0.704--Low set and/or angulated ears61.5% (16/26)57.9% (140/242)ND0.925--Ptosis37.5% (9/24)40.1% (97/242)9.3% (7/75)1.000 0.028
< 0.001 Hypertelorism50% (13/26)61.2% (148/242)52% (62/119)0.4701.0000.114Prominent and/or high forehead30.8% (8/26)10.7% (26/242)ND0.072--Triangle-shaped head26.9% (7/26)4.5% (11/242)ND 0.010 --Epicanthal folds23.1% (6/26)14.5% (35/242)ND1.000--Midface and/or malar hypoplasia23.1% (6/26)21.9% (53/242)ND0.469--Short stature23% (6/26)48.7% (134/275)17.4% (409/2346)0.0710.661 < 0.001 Macrocephaly42.3% (11/26)41.2% (93/226)31.3% (787/2516)10.443 0.003 Cardiovascular abnormalitiesCardiovascular malformations19.2% (5/26)36.8% (112/304)4% (121/3054)0.162 0.019
< 0.001 Pulmonic stenosis7.7% (2/26)24.3% (74/304)1.7% (61/3680)0.1610.181 < 0.001 Left heart obstruction (aortic stenosis/coarctation)3.8% (1/26)2% (6/304)0.3% (10/3849)0.6680.139 < 0.001 Mitral valves prolapse/dysplasia7.7% (2/26)5.6% (17/304)1.1% (36/3236)0.8110.119 < 0.001 Atrial septal defect0% (0/26)4.3% (13/304)0.3% (13/4646)0.8071 < 0.001 Ventricular septal defect0% (0/26)2% (6/304)0.3% (11/4338)11 < 0.001 Hypertrophic cardiomyopathy0% (0/26)2.6% (8/304)0.1% (4/2932)11 < 0.001 Electrocardiographic abnormality8.7% (2/23)0.7% (2/304)0.2% (5/2322)0.103 0.012 0.202VasculopathyCerebral vasculopathy3.8% (1/26)0% (0/305)4% (122/3024)0.16210Peripheral vasculopathy3.8% (1/26)0.6% (2/321)2.5% (2/77)0.37210.186Skin manifestationsCafé au lait spots (> 5)100% (26/26)96% (288/300)93.6% (3690/3943)0.8070.6610.112Skinfold freckling96.2% (25/26)64% (192/300)78.9% (3022/3831) 0.004 0.114 < 0.001 Superficial cutaneous neurofibromas (> 18 y)62.5% (5/8)37.6% (32/85)91% (803/882)0.4230.114 < 0.001 Subcutaneous neurofibromas (> 18 y)50% (4/8)14.1% (12/85)57.7% (297/515)0.1030.961 < 0.001 Plexiform neurofibromas (major external/severe; > 8 y)37.5% (3/8)12.6% (23/183)20% (169/847)0.1620.423 0.024 Neurological manifestationsLearning disabilities15.4% (4/26)32.8% (98/299)24.5% (260/1063)0.1620.497 0.005 Attention deficit hyperactivity disorders34.6% (9/26)7.4% (22/299)22.8% (227/996) 0.002 0.372 < 0.001 Clinical autism spectrum disorder0% (0/26)1% (3/299)7.8% (163/2077)10.473 < 0.001 Developmental delay/intellectual disability19.2% (5/26)25.8% (77/299)13.4% (154/1146)0.7120.632 < 0.001 Nervous-system tumoursOptic pathway gliomas (RMI and/or CT-scan; < 6 y)ND20% (4/20)18% (102/566)-0.793Malignant peripheral nerve sheat tumours3.8% (1/26)0% (0/321)3.4% (191/5682)0.1620.847 < 0.001 Lisch nodules (> 20 y)33.3% (1/3)51.4% (18/35)94% (102/108)^∃^10.062 < 0.001 Skeletal abnormalitiesScoliosis19.2% (5/26)23.2% (70/302)22.9% (240/1047)0.8110.9040.926Pectus excavatum61.5% (16/26)19.9% (57/287)1% (12/1157)^φ^ < 0.001
< 0.001
< 0.001 NF1 pathogenic variantTruncating34.6% (9/26)18.6% (54/280)54% (576/1067)0.1620.139 < 0.001 Missense38.5% (10/26)61.4% (172/280)9.2% (98/1067)0.102 < 0.001
< 0.001 Splice15.4% (4/26)3.2% (9/280)27.3% (291/1067)0.0890.390 < 0.001 In-frame0% (0/26)12.5% (35/280)2% (21/1067)0.1611 < 0.001 Large deletions11.5% (3/26)3.6% (10/280)7.5% (80/1067)0.1720.661 0.024 Abbreviations: CT, computed tomography; F, female; M, male; NF1, neurofibromatosis type 1; NS, Noonan syndrome; ND, not done; RMI, resonance magnetic imaging; y, years.
Facial phenotype analysis by clinicians
NSLFP was classified as ‘typical’ in 31% and ‘suggestive’ in 69% of cases. The inter-rater concordance showed substantial agreement, with a kappa of 0.65 [95% CI = 0.56; 0.74]; Suppl Fig. 1A). For sub-groups of patients aged under 12 years and 12 years or older, the inter-rater concordance showed moderate agreement, with kappa values of 0.47 [95% CI = 0.06; 0.88] and 0.73 [95% CI = 0.60; 0.87], respectively (Suppl Fig. 1B, 1 C). Common NSLFP features included low-set/angulated ears (61.5%), downslanted palpebral fissures (53.8%), hypertelorism (50%), and ptosis (37.5%). At least two of these anomalies were present in 100% of ‘typical’ cases and in 61% of ‘suggestive’ cases (Fig. 1).
Fig. 1. Neurofibromatosis-Noonan syndrome facial and thoracic features. (a) Prominent and high forehead, ptosis, hypertelorism, down-slanting palpebral fissures and low-set ears in a 5-year-old girl. (b and c) Hypertelorism, low set posteriorly angulated ears, high and broad nasal bridge, wide and prominent philtrum and triangular face in an 8-year-old girl. (d) Prominent and high forehead, high and broad nasal bridge, small and pointed chin in a 5-year-old-boy with recurrent p.Arg1809Cys NF1 pathogenic variant. (e) Low-set ears and prominent nasolabial folds in a 45-year-old woman. (f) Frontal bossing, hypertelorism and prominent nasolabial folds in a 45-year-old man. (g) Prominent and high forehead, high anterior hairline, low-set ears and bulbous nasal tip in a 27-year-old-woman. (h) Pectus excavatum and café-au-lait spots in an 8-year-old boy
Facial analysis by Face2Gene (F2G)
F2G ranked NS as the top match in 73% of cases and second match in 15% (Table S3). NF1 was listed in the top five in 88.5% and top ten in 100%. The most common alternative diagnoses included other RASopathies, like NS with multiple lentigines, cardiofaciocutaneous syndrome, NS-like disorders with loose anagen hair, and Costello syndrome.
Concordance between the clinicians and F2G was almost perfect with a kappa of 0.821 [CI 95% = 0.625; 1.000]) (Suppl Fig. 1D).
Cardiovascular malformations
Cardiovascular malformations were identified in 19.2% of cases, including pulmonic stenosis in 7.7%, with one case each of mild valvular and supravalvular stenosis, and mitral valve prolapse/dysplasia in 7.7%. Incomplete right bundle branch block was noted in 8.7%, and vasculopathies in 7.7%, including Moya-Moya disease with renal artery stenosis and ascending aortic dilatation.
Additional features
Key data are summarized in Table 1, with additional details in Table S4. All patients met NF1 diagnostic criteria and café-au-lait spots were universally present. Lentigines, superficial neurofibromas, subcutaneous neurofibromas, and plexiform neurofibromas were observed in 96.2%, 62.5%, 50% and 37.5%, respectively. Neurological manifestations were present in 61.5%, with attention deficit hyperactivity disorder (ADHD), developmental delay/intellectual disability, and learning disabilities observed in 34.6%, 19.2% and 15.4%, respectively. Macrocephaly, short stature, pectus excavatum and scoliosis were present in 42.3%, 23%, 62.5% and 19.2%, respectively.
Considering the diagnostic criteria of NS established by van der Burgt [62] and by Zenker [63], 38.5% and 30.8% of the patients, respectively, could also be diagnosed with NS. All identified NF1 PVs were classified as pathogenic/likely pathogenic and included missense, truncating, splice and large deletions, occurring in 38.5%, 34.6%, 15.4% and 11.5%, respectively. Of the 10 missense PVs, 80% were previously reported as being associated with NF-NS, specifically NF1 p.(Arg1809) (50%, 2 families), p.(Arg1276) (20%, 2 families) and p.(Lys1423) (10%) No PVs were found in other RASopathies-related genes.
Comparison with ‘classic’ NF1 and literature
A literature review of 321 cases NF-NS (Table S1) [1–52] found that 94.7% met NIH NF1criteria. The most common NSLFP features were hypertelorism (61.2%), low-set and/or angulated ears (57.9%), downslanted palpebral fissures (45.9%), and ptosis (40.1%). At least two of these four anomalies were present in 70.7% of the cases. Cardiovascular malformations were noticed in 36.8% of cases, including pulmonic stenosis in two-thirds. Scoliosis and pectus excavatum were observed in 23.2% and 19.9%, respectively.
PVs in the NF1 gene were identified in 87.2% of cases, with missense, truncating, in-frame, large deletions, and splice variants found in 61.4%, 18.6%, 12.5%, 3.6% and 3.2%, respectively. Additionally, co-occurring PVs in other RASopathies-associated genes were noted in eight cases, including seven with PTPN11 PVs [8, 14, 29, 33, 44] and one with a KRAS PV [5]. Overall, PTPN11 PVs were identified in 4.7% of cases.
Discussion
Our study confirms that NF-NS is a rare phenotypic variant of NF1, with a frequency of 4.7% in our cohort, consistent with the literature reports ranging from 2% to 6.4% [4, 64–67]. However, these findings are often heterogeneous due to the lack of standardized diagnostic criteria for NF-NS. We included patients with NF1 confirmed by NIH criteria and molecular analysis of NF1 gene, who exhibited typical or suggestive facial abnormalities (“gestalt”) of NS. Molecular confirmation of the NF1 was essential to avoid misdiagnosing NF-NS as other RASopathies with overlapping features, such as café-au-lait spots and lentigines, seen in Legius syndrome [67], NS [68], NS with multiple lentigines [68], and heterozygous LZTR1 variants [69]. At inclusion, we did not consider other NS diagnostic criteria (e.g., short stature, thoracic or cardiac malformations) but focused on NSLFP as the cornerstone of NF-NS diagnosis due to its clinical relevance and lower susceptibility to bias. NF1 lacks a distinctive facial phenotype among RASopathies [57, 70], and short stature, a common feature in 20% of ‘classic’ NF1 [14, 32, 65, 66, 71–75], lacks discriminatory value. Similarly, in ‘classic’ NF1, pectus deformities remained underexplored [76], while congenital cardiovascular malformations have been reported with frequencies ranging from 0.4 to 8.6% [74], with PVS present in 1.7% [12, 32, 77–81]. Studies on PVS are limited by small sample sizes and depend on whether the diagnosis was based on or confirmed by auscultation or echocardiography [81].
Recognizing NSLFP is challenging, as features evolve and become more subtle with age [82]. Inter-rater agreement among clinicians was moderate (κ = 0.65 [95% CI = 0.56; 0.74]) reflecting the inherent variability and subjectivity in assessing facial phenotypes. The presence of two or more facial abnormalities (e.g., low-set and/or angulated ears, downslanted palpebral fissures, hypertelorism, and ptosis) is a valuable diagnostic indicator, consistently observed in ‘typical’ NF-NS cases. According to the literature, these features are noticed in nearly three-quarters of cases. However, the specificity of these features in ‘classic’ NF1 remains undetermined in the absence of dedicated studies. Hypertelorism and ptosis have been reported in 52% [72, 83] and 9.3% [84] of ‘classic’ NF1 cases, but the small number of observations precludes definitive conclusions.
F2G analysis demonstrated high performance, ranking NS as the top match in 73% of cases and NF1 among the top five in 88.5%. Despite relying solely on front-facing images, F2G’s performance was comparable to clinicians with access to comprehensive data. Concordance between F2G and clinicians in identifying typical or suggestive NSLFP was near-perfect (κ = 0.821). While NF1 has been historically thought to lack distinct facial features, recent studies using deep learning suggest subtle facial characteristics in NF1 compared to controls [85], or within RASopathies, particularly milder CS features [70]. These technologies have limitations, including population-specific traits [85], but their precision could improve with the inclusion of clinical data or genetic information.
Our study detailed anthropometric, dermatological, neurological, ocular, and skeletal findings, which were broadly consistent with the literature. We observed a higher frequency of skinfold freckling (96.2% vs. 64%) and ADHD (34.9% vs. 7.4%), likely due to systematic data collection and evolving diagnostic criteria for ADHD. Pectus excavatum was present in 61.5%, higher than the 19.9% reported in the NF-NS literature, possibly due to our inclusion of minor cases. Aside from NSLFP and pectus excavatum, our study did not identify a distinct phenotype compared to ‘classic’ NF1, aligning with a previous detailed series of 22 NF-NS patients [16] and contrasting with other studies focusing on specific NF1 patients with NF1 PVs [22–24, 31, 38, 45] or associated CHM [32].
CHM in ’classic’ NF1 is reported in 4% of cases, ranging from 0,4 to 6,4% with PVS occurring in 1.7% overall [12, 77–81, 86]. This may be underestimated as cardiovascular assessments often rely on auscultation [81]. Echocardiography, routinely recommended for other RASopathies [53], is not yet established for ‘classic’ NF1. The association between NF-NS and higher CHD risk, including PVS, and prevalent missense or in-frame NF1 PVs [32], such as p.(Arg1276) [24], p.(Lys1423) [24], and p.(Arg1809) [31], is supported by pooled literature data. More globally, the increased risk of CHM in NF-NS, regardless of the type of NF1 pathogenic variant, appears to be confirmed by pooled data from the literature, with a significantly increased frequency of CHM and PVS at 36.8% and 24.3%, respectively. Our study, conducted without presupposing NF1 PV types, confirmed an increased CHM risk (19%), with a trend toward increased PVS (7.7%), left heart obstruction (3.8%), and mitral valve prolapse/dysplasia (7.7%). These findings support the need for an initial cardiac evaluation, including echocardiography, in all NF1 patients with NSLFP, regardless of the type of NF1 PVs. Furthermore, given the potential for late-onset or progressive cardiac manifestations, we recommend periodic cardiac follow-up over time, similar to surveillance guidelines in RASopathies, even in the absence of initial cardiological abnormalities”.
NF1 PVs remain the primary molecular event underlying NF-NS. In our cohort, the frequency of truncating PVs was higher than reported in the literature; however, we also observed a high frequency of recurrent missense PVs in 38.5%, consistent with previous findings. Additionally, RASopathy PVs, mostly linked to the PTPN11, were observed at a frequency of 5.7% based on pooled data [5, 8, 14, 29, 33, 44]. A recent study reported PTPN11 PV in 2.9% of NF1 patients, 75% of whom exhibited an NS-like phenotype [14]. Although we did not identify co-occurring RASopathy PVs in our cohort, possibly due to the limited sample size, these findings support systematic screening for RASopathy PVs in NF1 patients who exhibited an NS-like phenotype.
Conclusions
This study highlights that NF-NS is a distinct phenotypic variant of NF1, confirmed through both molecular and clinical analyses. Future advancements in facial phenotype analysis, particularly deep-learning technologies, offer promising tools for helping clinicians diagnose NF-NS earlier and more accurately. Given the increased prevalence of CHM, our findings suggest that early recognition of NSLFP in NF1 patients should prompt a more proactive cardiovascular evaluation. The frequent association of NF-NS with missense and in-frame PVs in the NF1 gene, as well as the rare but significant co-occurrence of RASopathy PVs, underscores the importance of systematic RASopathy variant testing and genetic screening in this population.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Material 1
Supplementary Material 2
Supplementary Material 3
Supplementary Material 4
Supplementary Material 5
Supplementary Material 6
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Neurofibromatosis conference statement. National Institutes of Health Consensus Development Conference. Arch. Neurol. 1988;45:575–8.3128965 · pubmed ↗
- 2Mastromoro G, Santoro C, Motta M, Sorrentino U, Daniele P, Peduto C, et al. Heterozygosity for loss-of-function variants in LZTR 1 is associated with isolated multiple cafe-au-lait macules. Genet Med. 2024 Aug;10:101241.10.1016/j.gim.2024.10124139140257 · doi ↗ · pubmed ↗
- 3Matthews H, Vanneste M, Katsura K, Aponte D, Patton M, Hammond P, et al. Refining nosology by modelling variation among facial phenotypes: the rasopathies. J Med Genet. 2022 Jul;20:jmedgenet–2021.10.1136/jmedgenet-2021-108366 PMC 985236135858754 · doi ↗ · pubmed ↗