Location and extent of disease predicts outcome of neurofibromatosis type 1-related pediatric low-grade gliomas
Sneha M Chaturvedi, Arohi Saxena, Ahmad Hassan, Ali Mian, Chelsea Kotch, Nicole M Brossier

Abstract
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
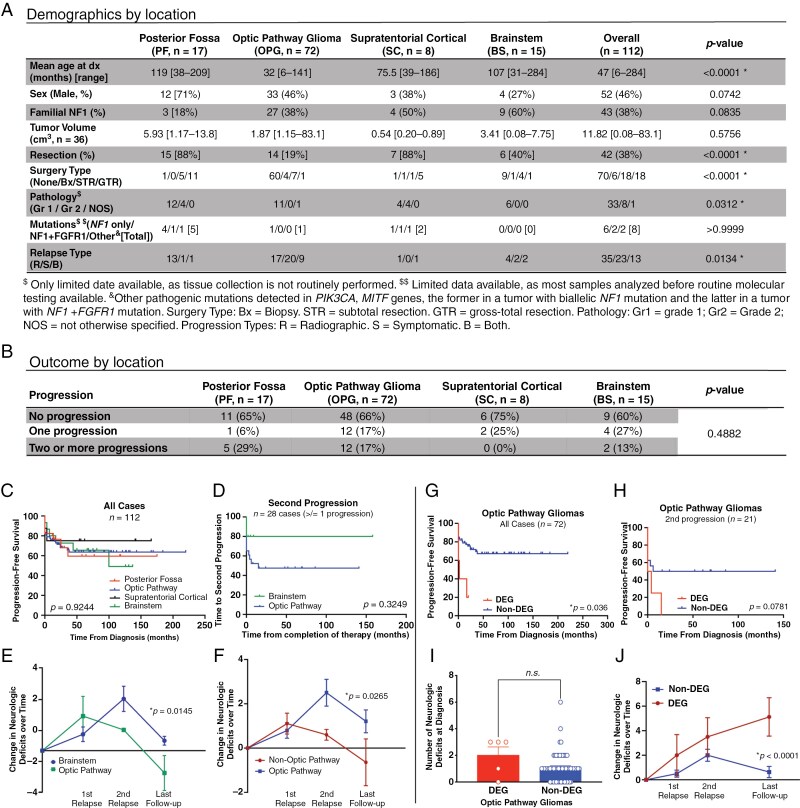 Figure 1
Figure 1- —Neurofibromatosis Therapeutic Acceleration Program10.13039/100014804
- —St. Louis Children’s Hospital
- —Neurofibromatosis Therapeutic Acceleration Program10.13039/100014804
- —National Institutes of Health10.13039/100000002
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsNeurofibromatosis and Schwannoma Cases · Glioma Diagnosis and Treatment · Neuroblastoma Research and Treatments
Children with neurofibromatosis type 1 (NF1) are at increased risk of developing low-grade gliomas (LGGs) in multiple brain locations.^1^ In children with sporadic pilocytic astrocytoma, location impacts the rate of progression and accumulation of neurological deficits,^2^ with supratentorial midline (SM, including optic pathway) tumors exhibiting worse neurologic morbidity over time. We performed a multi-institutional retrospective cohort study to determine how location affects progression and functional morbidity in NF1-LGG. We observed an increased risk of symptomatic relapse and more neurologic deficits over time in optic pathway tumors compared to other locations; this risk was most pronounced in children with deep extensive gliomas (DEGs).
Children with NF1 are at risk for the development of LGG, which arises most commonly in the optic pathway (optic pathway glioma: OPG) and brainstem (BS).^3,4^ When symptomatic, OPG typically manifests with visual deficits or endocrinopathies, while LGG in other locations may present with headache, seizure, weakness, or other neurologic impairments.^1,3–5^ One-third to one-half of NF1-LGG require treatment, including surgical resection and/or chemotherapy.^6^ Studies in sporadic gliomas have found that tumor location is associated with outcomes such as tumor progression and functional deficit burden.^2,7^ However, it is unclear whether tumor location is similarly associated with outcomes in NF1 patients, who often have a more indolent clinical course than individuals with sporadic LGG. For these reasons, we conducted a multi-center retrospective cohort study to evaluate the impact of tumor location on long-term outcomes for children with NF1-LGG.
This multi-institutional retrospective chart review received approval from the institutional review boards at St. Louis Children’s Hospital and Children’s Hospital of Philadelphia. The study population included 99 children 18 years of age or younger at the time of initial diagnosis of NF1-LGG who received treatment for their target tumor between 1999 and 2021. Per consensus recommendations for NF1-associated glioma,^8^ tumors were classified as probable LGG by radiographic features; biopsies were not standardly performed. Patients with multiple gliomas in distinct locations had each tumor counted separately, with 112 total NF1-LGG included in this study. Each NF1-LGG was categorized into one of 4 locations: posterior fossa (PF, n = 17, 15%; not including brainstem), optic pathway (OPG, n = 72, 64%, including the optic nerves, chiasm, radiations, and hypothalamus), supratentorial cortical (SC, n = 8, 7%), and brainstem (BSG, n = 15, 13%) (Figure 1A). OPGs were further classified into DEGs or non-DEGs, with DEGs defined as tumors involving the optic pathway and bilateral temporal lobes, thalami and basal ganglia.^1^ Tumor progression was determined based on radiographic or symptomatic criteria. Neurologic deficits (described in the figure legend) were determined at baseline, each progression, and at the last follow-up.
In the analytic cohort, OPGs presented at a younger age compared to PF tumors (32 vs. 119 months; Figure 1A). No significant association was identified between tumor location and biological sex or familial NF1 status. PF tumors were significantly more likely to undergo surgical intervention than OPGs (88% vs. 19%). OPGs displayed higher rates of symptomatic relapses, such as visual acuity decline, when compared against all non-OPG tumors (63% vs. 39%).
There was no association between tumor location and the number of progressions over time (Figure 1B) or the time to first progression (Figure 1C). These data contrast with sporadic tumors, where SM and BS pilocytic astrocytoma have shorter progression-free survival (PFS) due to higher rates of subtotal resection.^2^ Additional analyses comparing OPG versus BSG subgroups were performed, given higher rates of treatment of NF1-LGG in these locations.^1,3,4^ Mean longitudinal follow-up was similar between the 2 (77 vs. 79 months, respectively). OPGs trended toward a shorter time to second progression when compared to the BSG subgroup, but this did not reach statistical significance, likely due to insufficient sample size (Figure 1D). OPGs showed more neurologic deficits over time than BSGs (Figure 1E) and all non-OPGs (interaction, p = 0.0071; not shown). In patients that had undergone at least one relapse, OPGs also showed a trend toward more severe neurologic deficits over time than BSGs (not shown) and a statistically significant increase in neurologic deficits over time when compared to all non-OPGs (Figure 1F). From the entire patient cohort, one patient with BSG died before the end of the study period, consistent with previously described overall survival rates for patients with NF1-LGG.^9^
NF1-related DEGs have been associated with poorer PFS.^1^ We evaluated the OPG group by the extent of disease, dividing patients into those that met the criteria for DEG (n = 5) versus those that did not (n = 67). All DEGs were infiltrative across multiple adjacent brain regions at first treatment, although not all met the criteria for DEG at that time. All DEG tumors were identified in males, a sex bias not seen previously. DEGs demonstrated a significantly shorter PFS (Figure 1G), which aligns with previous studies.^1^ Four of the 5 patients with DEG demonstrated at least 2 progression events, with a median time to second progression of 4.75 months (range 0–15). There was a trend toward shorter PFS at the second progression than non-DEG (Figure 1H; p = 0.0781). DEGs did not have a greater number of neurologic deficits at diagnosis (Figure 1I) but were associated with increased accumulation of neurologic burden over time (Figure 1J).
This analysis had several limitations, including sample size, abstraction of progression and neurologic deficits by retrospective chart review, the inclusion of both symptomatic and asymptomatic (radiographic) progressions, and comparatively few progressive tumors identified outside the optic pathway. Despite these limitations, our study identified a higher rate of symptomatic disease progression and poorer longitudinal outcomes in individuals with NF1-OPG compared to NF1-LGG in other locations, confirming the SM location as a risk factor for poorer disease outcomes in NF1-LGG. Within the OPG cohort, DEGs appear to be at particularly increased risk for recurrent progression and increased number of neurologic deficits over time, confirming the extent of tumor infiltration as a clinical feature associated with tumor progression^10^ and suggesting that these patients in particular may benefit from alternative treatment strategies. Prospective studies, such as the ongoing NF1-LGG and NF1-OPG natural history studies, will be needed to confirm tumor location and extent of disease as prognostic factors in NF1-LGG.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Mahdi J , Goyal MS, Griffith J, Morris SM, Gutmann DH. Nonoptic pathway tumors in children with neurofibromatosis type 1. Neurology.2020;95(8):e 1052–e 1059.32300062 10.1212/WNL.0000000000009458 PMC 7668552 · doi ↗ · pubmed ↗
- 2Brossier NM , Strahle JM, Cler SJ, Wallendorf M, Gutmann DH. Children with supratentorial midline pilocytic astrocytomas exhibit multiple progressions and acquisition of neurologic deficits over time. Neurooncol. Adv..2021;4(1):vdab 187.35036914 10.1093/noajnl/vdab 187PMC 8757579 · doi ↗ · pubmed ↗
- 3Listernick R , Charrow J, Greenwald MJ, Esterly NB. Optic gliomas in children with neurofibromatosis type 1. J Pediatr.1989;114(5):788–792.2497236 10.1016/s 0022-3476(89)80137-4 · doi ↗ · pubmed ↗
- 4Mahdi J , Shah AC, Sato A, et al A multi-institutional study of brainstem gliomas in children with neurofibromatosis type 1. Neurology.2017;88(16):1584–1589.28330960 10.1212/WNL.0000000000003881 PMC 5395076 · doi ↗ · pubmed ↗
- 5Habiby R , Silverman B, Listernick R, Charrow J. Precocious puberty in children with neurofibromatosis type 1. J Pediatr.1995;126(3):364–367.7869193 10.1016/s 0022-3476(95)70449-3 · doi ↗ · pubmed ↗
- 6Campen CJ , Gutmann DH. Optic pathway gliomas in neurofibromatosis type 1. J Child Neurol.2018;33(1):73–81.29246098 10.1177/0883073817739509 PMC 5739070 · doi ↗ · pubmed ↗
- 7Fernandez C , Figarella-Branger D, Girard N, et al Pilocytic astrocytomas in children: prognostic factors—a retrospective study of 80 cases. Neurosurgery.2003;53(3):544–553; discussion 554.12943571 10.1227/01.neu.0000079330.01541.6e · doi ↗ · pubmed ↗
- 8Packer RJ , Iavarone A, Jones DTW, et al Implications of new understandings of gliomas in children and adults with NF 1: report of a consensus conference. Neuro Oncol. 2020;22(6):773–784.32055852 10.1093/neuonc/noaa 036PMC 7283027 · doi ↗ · pubmed ↗