Genetic Terminal Complement Deficiency in Israeli Bedouins With Kidney Failure
Guy Chowers, Dror Ben-Ruby, Danit Atias-Varon, Omer Shlomovitz, Keren Slabodnik-Kaner, Maayan Kagan, Shany Avayou, Elvira Romanjuk, Boris Rogachev, Yosef S. Haviv, Ohad S. Birk, Noam Hadar, Younes Bathish, Iris Barshack, Alexander Volkov, Camila Avivi, Anna Pavlovsky

Abstract
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
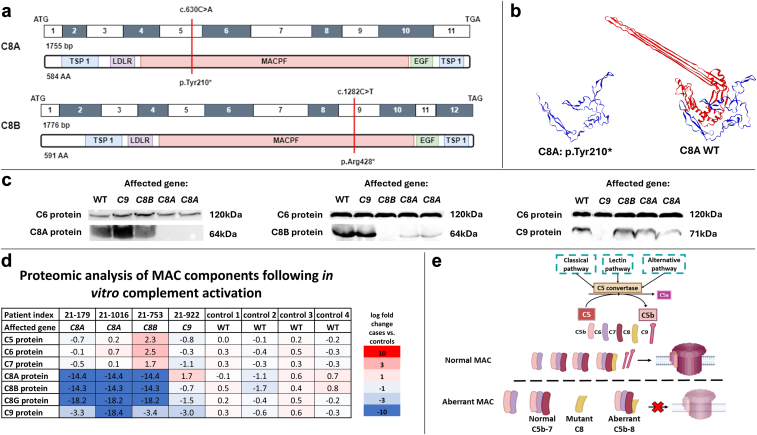 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRenal Diseases and Glomerulopathies · Hemoglobinopathies and Related Disorders · Erythropoietin and Anemia Treatment
Introduction
The prevalence of chronic kidney disease (CKD) is particularly high among Israeli non-Jewish populations,S1^,^S2 often attributed to genetic kidney diseases.1 We established the Israeli Kidney Failure Genetic Cohort to reveal the genetic causes of kidney failure in adults across diverse ethnicities.1 Here, we studied the Israeli Bedouins, a seminomadic, Arabic-speaking minority. The Bedouin social structure is based on nuclear families integrated into tribes with high endogamy and consanguinity rates nearing 45%.2^,^S3^,^S4 The Bedouin population primarily resides in North Africa, the Sinai and Arabian Peninsulas, as well as in Israel's southern Negev desert and Galilee region. In this pilot study, we aimed to uncover the genetic landscape of kidney failure in Israeli Bedouins by focusing on identifying high-penetrance monogenic causes using exome sequencing (ES).
Results
Clinical characteristics of the study population (N = 81) are presented in Supplementary Table S1. Using ES, we identified genetic causes in 10 participants (12%), including 6 distinct monogenic disorders (Table 1).3^,^S5-S10 The mean age at kidney failure diagnosis, along with rates of parental consanguinity and family history of kidney failure were similar for patients with genetically driven kidney failure compared to patients with negative ES. Pathogenic COL4A3 variants were detected in 5 participants, making COL4A3 variants the most common molecular genetic diagnosis in our cohort (Supplementary Figure S1). Genetic diagnosis was often discordant with the initial clinical diagnosis and influenced subsequent clinical management (Supplementary Table S2).Table 1. Clinical data and post–exome sequencing molecular-genetic diagnoses in 13 Bedouin individuals in whom diagnostic variants were identifiedPatientPre–exome sequencing clinical diagnosisFHxPost–exome sequencing diagnosisGeneAnnotationZygosityCADD scoreaAF gnomADbAF in matched controls (n = 399)cACMGdACMG criteriadMonogenic kidney diseases21–759Male with ADPKD, hearing loss; kidney failure at age of 54 yrsYesType 4 collagen related CKD (Alport syndrome spectrum)COL4A3c.361G>A, p.Gly121SerHom25.20.0009%1∖798 0.13%LPPM2 (Supp), PM1, PP3 (Mod), PM3 (Supp)21–1209Male with diabetic nephropathy; kidney failure at age of 64 yrsYesType 4 collagen related CKD (Alport syndrome spectrum)COL4A3c.361G>Ap.Gly121SerHet25.20.0009%1∖798 0.13%LPPM2 (Supp), PM1, PP3 (Mod), PM3 (Supp)21–990Male with nephropathy of unknown cause; kidney failure at age of 35 yrsYesType 4 collagen related CKD (Alport syndrome spectrum)COL4A3c.2275G>C, p.Gly759ArgHet390.0001%3∖798 0.38%LPPP3 (Mod), PM1, PM2 (Supp), PS1 (Supp),21–1614Female with nephropathy of unknown cause; kidney failure at age of 54 yrsNoType 4 collagen related CKD (Alport syndrome spectrum)COL4A3c.2275G>C, p.Gly759ArgHet390.0001%3∖798 0.38%LPPP3 (Mod), PM1, PM2 (Supp), PS1 (Supp),21–715Female with diabetic nephropathy; kidney failure at age of 73 yrsNoType 4 collagen related CKD (Alport syndrome spectrum)COL4A3c.3829G>Tp.Gly1277CysHet25.80.0%1∖798 0.13%LPPP3 (Mod), PM2 (Supp), PM5, PP221–755Female with GSD type 1b, hearing loss, and intellectual disability; kidney failure at age of 20 yrs; parental consanguinityNoGlycogen storage disease type 1bSLC37Ac.1042_1043delCT p.Leu348fsHom350.018%0/7980%PPVS1, PM2 (Supp), PS421–1028Female with FSGS, hypothyroidism; kidney failure at age of 20 yrsNoPapillorenal syndromePAX2c.76dupGp.Val26fsHet370.0005%0/7980%PPVS1, PM2 (Supp), PS221–1064Male with nephropathy of unknown cause, heart failure; kidney failure at age of 20 yrs; parental consanguinityNoNephronophthisis 4NPHP4chr1:g.5964667_6008322delHomN/AeN/AN/AP2E, 4L21–1210Female with hemolytic uremic syndrome; kidney failure at age of 52 yrsYesAtypical HUSCFHc.3493+5G>AHet22.60.0%0/7980%LPfPP3, PS1 (Mod), PM2 (Supp), PS4 (Mod), PP421–1311Male with ADPKD; kidney failure at age of 48 yrsYesADPKDPKD2c.1390C>Tp.Arg464∗Het370.0%0/7980%PPVS1, PM2 (Supp), PS4Other monogenic diseases (terminal complement)21–179Male with IgA nephropathy; kidney failure at age of 13 yrs; parental consanguinityNoTerminal complement deficiencyC8Ac.630C>Ap.Tyr210∗Hom410.0008%11/7981.38%PPVS1, PM2 (Supp), PS321–1016Male with IgA nephropathy; kidney failure at age of 13 yrs; parental consanguinityNoTerminal complement deficiencyC8Ac.630C>Ap.Tyr210∗Hom410.0008%11/7981.38%PPVS1, PM2 (Supp), PS321–753Male with diabetic nephropathy; kidney failure at age of 32 yrs; parental consanguinityNoTerminal complement deficiencyC8Bc.1282C>Tp.Arg428∗Hom390.13%2/7980.25%PPVS1, PM2 (Supp), PS3ACMG, American College of Medical Genetics classification; ADPKD, autosomal dominant polycystic kidney disease; AF, allele frequency; B, benign; CADD score, combined annotation dependent depletion score; CAKUT, congenital anomalies of the kidney and urinary tract; CKD, chronic kidney disease; DM, diabetes mellitus; FHx, family history of kidney disease; FSGS, focal segmental glomerulosclerosis; gnomAD, genome aggregation database; GSD, glycogen storage disease; Het, heterozygous; Hom, homozygous; HUS, hemolytic uremic syndrome; LB, likely benign; LP, likely pathogenic; Mod, moderate; P, pathogenic; Supp, supporting; VUS, variant of unknown significance.Only 1 patient had a prior molecular diagnosis (glycogen storage disease type 1b) and 2 participants had a clinical phenotype-based suspicion of ADPKD; One participant was confirmed to have a pathogenic PKD2 variant, the other participant had a homozygous COL4A3 variant classified as likely pathogenic. COL4A pathogenic variants have recently been reported as a cause of cystic kidney disease, which can mimic ADPKD.3^,^S5^,^S6 A manual inspection of PKD1 and PKD2 to verify sequencing adequacy was conducted, confirming 98% and 100% of regions covered at >20x depth, respectively, thus minimizing the likelihood of undetected pathogenic variants in these genes.aIn silico prediction tool for scoring the deleteriousness of variants, higher values of CADD score predict a more severe effect on a logarithmic scale.S7bAccording to the gnomAD version 4.1.0.S8cIn-house controls include 399 Bedouins from the Negev region in Israel.dACMG: according to the ACMG classification, a variant can be classified as P, LP, VUS, LB; or B. All variant classified with the https://franklin.genoox.com platform. The level of evidence was assigned based on the ACMG guidelines and the updated ClinGen SVI recommendations. Any adjustments made to these default classifications are indicated in brackets.eNeither the exact deletion nor a similar deletion with > 50% overlap were found using CADD-SV tool.S7fApplication of the PS4 criterion based on previous reports of this variant in two affected cases and absence in controls.S9 PP3 and PS1 (Mod) used based on ClinGen SVI guidelines for splice variants.S10
Terminal Complement Pathogenic Variants Among Bedouins With Kidney Failure
Three unrelated patients were found to harbor nonsense variants in 2 terminal complement genes, 1 patient with C8B variant and 2 patients with C8A variant (Supplementary Figure S1, Table 13^,^S5-S10). These genes translate to the α and β chains of complement factor 8, which comprise the membrane attack complex (MAC)—together with C5-9—the final common pathway of the complement cascade. These 2 variants have been previously reported in the context of C8 deficiency, which causes susceptibility to encapsulated infections.4^,^5 Both truncating variants are located within the MAC/perforin domain (Figure 1a and b).Figure 1. Identification of variants in C8A and C8B in 3 Israeli Bedouin individuals with kidney failure. (a) Exon structure of human C8A and C8B cDNA and domain structure of complement factor 8 alpha and beta chains (C8A and C8B). Nucleotide and protein changes are highlighted by red lines. Both variants were located in the MACPF domain. This domain is crucial for target membrane insertion and the formation of a transmembrane channel that disrupts the cell, causing lysis. ATG and TGA/TAG stop codons are indicated. (b) 3D model of C8A with and without the truncating variant. The model was generated the SWISS-MODEL.S16 The N terminus upstream of the early stop codon at residue 210 is shown in blue. The C terminus, downstream of the early stop codon, is shown in red. (c) Western blot of serum from patients with deficits in terminal complement proteins. When probing the C8B chain, in addition to the anticipated protein absence in the case of the C8B variant, the 2 cases with the C8A variant showed reduced protein levels. The label in each lane indicates the affected gene in each patient, including 1 healthy, unrelated individual as a control (labeled WT). C6 was used as the loading control. (d) Proteomic analysis of the MAC components showing aberrant MAC formation. Complexes were analyzed following in vitro complement activation and pull-down of C6 containing complexes using co-IP. Each sample was compared to the average protein levels of controls. Whereas complete C5b-9 MAC were formed in controls, very low levels of C8 and C9 were detected in the cases. (e) Normal complement cascade and suggested pathological mechanisms leading to kidney disease. Abnormal C8 or C9 proteins result in the inability of C9 monomers to connect to the forming MAC and the absence of intact membranes penetrating pores. The resulting partial MAC, lacking C9 and some C8 chains, may lead to complement deposition in the glomeruli, either by itself as protein aggregates or as a part of immune complexes. co-IP, coimmunoprecipitation; EGF, epidermal growth factor like domain; LDLR, low density lipoprotein receptor class A domain; MAC, membrane attack complex; MACPF, membrane attack complex/perforin domain; TSP1, thrombospondin 1 domain.
Patient 21-179 with a C8A pathogenic variant developed kidney failure at the age of 13 years. A renal biopsy revealed mesangial IgA deposits (Supplementary Figure S2, Table 13^,^S5-S10). The patient experienced rapid progression despite treatment and eventually required dialysis. Subsequently, he underwent 2 kidney transplantations, which were lost because of acute rejection attributed to thrombotic microangiopathy and recurrence of IgA and C3 deposits, respectively. Similarly, patient 21-1016 presented at age 9 with elevated creatinine levels and a kidney histopathological pattern of IgA nephropathy. He progressed to kidney failure by the age of 13 years and required 3 transplants over the next 20 years because of recurrent rejection. Patient 21-753 with C8B variant presented with type 2 diabetes mellitus and progressive proteinuria, leading to kidney failure at age of 32 years. Unfortunately, renal biopsy was not performed. None of the patients had a history of infection before or after the onset of kidney disease.
C8 deficiency has not previously been associated with kidney disease. However, the C8A variant was significantly enriched in our cohort compared to ethnically matched controls. The homozygous genotype was enriched among cases compared with controls (P = 0.04, Fisher exact test). The minor allele frequency was higher in cases than in controls (3.7% vs. 1.38%, P = 0.04). This enrichment was driven by a higher proportion of homozygotes (2/81 vs. 0/399, P = 0.002) rather than by differences in carrier rates (2/81 vs. 11/399, P = 0.9). This enrichment led us to screen for variants in the terminal complement genes among 1300 Israeli patients with kidney failure. Consequently, we identified an Arab-Israeli patient with a likely pathogenic homozygous variant in C9 (c.355T>G, p.Cys119Gly). Unfortunately, this patient did not undergo renal biopsy.
Terminal Complement Variants Result in Aberrant MAC Formation
To study the association between terminal complement variants and kidney injury, we examined the deleterious effects of these variants. We measured the presence of each component of the complement cascade in the patients’ serum and total hemolytic activity using the CH50 assay (Supplementary Table S3). Reduced levels of C8 and C9 proteins were observed in 4 participants harboring C8A, C8B, or C9 pathogenic variants, respectively. Accordingly, all 4 cases had low CH50 levels, indicating an impaired ability to form a functional MAC. The other components of the complement system were not remarkably altered (Supplementary Table S3). Western blot analysis showed the absence of the corresponding proteins in all cases, consistent with the identified pathogenic variants (Figure 1c).
We hypothesized that the formation of aberrant MACs due to the inability to connect C9 monomers to the initial C5b-7 complex, may contribute to kidney disease. To detect these partial complexes following complement activation, we performed coimmunoprecipitation, followed by mass spectrometry (Figure 1d and e). Mass spectrometry revealed that in patient sera, incomplete MACs were formed and composed only of C5b-7 components (or C5b-8 in the case of the C9 variant). This suggests that following complement activation, aberrant MACs are formed in affected patients, which may subsequently result in uncleared glomerular depositions.
Discussion
In this nationwide study, exome sequencing of 81 Israeli Bedouins with kidney failure identified diagnostic variants in 12% of cases that were often clinically undiagnosed. In addition, we identified a cluster of monogenic terminal-complement deficiencies, suggesting a potential novel link to CKD.
Our findings provide several insights. First, the incidence of monogenic kidney disease among the Bedouin population is higher than previously reported in heterogeneous populations of European ancestry.6 Given the underrepresentation of Bedouins in genetic databases, our findings underscore the need for more diverse international study cohorts. Second, the low rate of previous genetic diagnoses and poor concordance with pre-exome diagnoses highlights the genetic and phenotypic heterogeneity of CKD. Furthermore, in contrast to previous studies suggesting that PKD1 and PKD2 are the most common genetic causes of kidney failure,6^,^7 our findings indicate that among Bedouins, COL4A3 variants are the predominant genetic etiology. Consistent with previous studies,1^,^S11 our findings suggest that genetic CKD is characterized by a population-specific genetic landscape.
Finally, our results support a possible emerging role of aberrant terminal complement activity in CKD. Strikingly, 2 patients with C8A variants were diagnosed with kidney failure due to IgA nephropathy, requiring kidney replacement therapy by the age of 13 years. This extremely rare childhood phenotype suggests a potential contributory role of C8A pathogenic variants. Whereas research on complement dysregulation in kidney disease has focused on early cascade components, the role of terminal complement, particularly its link to CKD and IgA nephropathy, remains less understood, with associations to renal disease being inconsistent.8^,^9^,^S12 A French cohort study reported noninfectious or autoimmune diseases, including kidney disease, in 16 of 154 patients with terminal complement deficiencies,4 whereas in a UK cohort of 40 such patients kidney involvement was not reported.5 Notably, C8 and C9 knockout murine models have not been assessed for kidney injury.S13^,^S14
Our results indicated reduced functional complement activity in all 4 patients with terminal complement variants. We hypothesize that this deficiency contributes to kidney disease via the formation of incomplete MAC deposits in the glomeruli, either independently or within immune complexes. It is plausible that such deposition may require a “second hit” trigger, like an infection, to activate the complement system, potentially exacerbating existing conditions.8^,^9 Alternatively, impaired MAC formation might lead to overactivation of early complement components, a mechanism linked previously to C3 glomerulopathy and membranoproliferative glomerulonephritis,8 or defective clearance of apoptotic cells, possibly triggering autoimmunity.S15 In summary, the high prevalence of terminal complement deficiencies in our cohort suggests a possible role in kidney disease. These deficiencies may act as primary causes of kidney disease or as exacerbating factors in various kidney pathologies, possibly mediated by the formation of aberrant MACs. These insights may have clinical implications, both for precise genetic diagnosis and possible employment of available complement targeting therapies. Further studies with larger and more diverse cohorts are needed to better understand the underlying molecular mechanisms.
Disclosure
The authors declare that they have nothing to disclose.
Patient Consent
Written informed consent was obtained from all participants.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Shlomovitz O.Atias-Varon D.Yagel D.Genetic markers among the Israeli Druze minority population with end-stage kidney disease Am J Kidney Dis 83202418319510.1053/j.ajkd.2023.06.00637717846 · doi ↗ · pubmed ↗
- 2Singer S.Davidovitch N.Abu Fraiha Y.Abu Freha N.Consanguinity and genetic diseases among the Bedouin population in the Negev J Community Genet 112020131910.1007/s 12687-019-00433-831376120 PMC 6962410 · doi ↗ · pubmed ↗
- 3Bada-Bosch T.Sevillano A.M.Sanchez-Calvin M.T.Cystic phenotype and chronic kidney disease in autosomal dominant Alport syndrome Nephrol Dial Transplant 3920241288129810.1093/ndt/gfae 00238178635 · doi ↗ · pubmed ↗
- 4El Sissy C.Rosain J.Vieira-Martins P.Clinical and genetic spectrum of a large cohort with total and sub-total complement deficiencies Front Immunol 102019193610.3389/fimmu.2019.0193631440263 PMC 6694794 · doi ↗ · pubmed ↗
- 5Shears A.Steele C.Craig J.Clinical outcome and underlying genetic cause of functional terminal complement pathway deficiencies in a multicenter UK cohort J Clin Immunol 42202266567110.1007/s 10875-022-01213-935084692 PMC 8793329 · doi ↗ · pubmed ↗
- 6Groopman E.E.Marasa M.Cameron-Christie S.Diagnostic utility of exome sequencing for kidney disease N Engl J Med 380201914215110.1056/NEJ Moa 180689130586318 PMC 6510541 · doi ↗ · pubmed ↗
- 7Vivante A.Genetics of chronic kidney disease N Engl J Med 391202462763910.1056/NEJ Mra 230857739141855 · doi ↗ · pubmed ↗
- 8Petr V.Thurman J.M.The role of complement in kidney disease Nat Rev Nephrol 19202377178710.1038/s 41581-023-00766-137735215 · doi ↗ · pubmed ↗