Genomic Characterization of Potential Opportunistic Zoonotic Streptococcus parasuis Isolated in China
Gang Liu, Yu Liu, Zhikang Jiang, Kang Liu, Xianwen Wang, Juyuan Hao, He Kong, Yajie Yu, Zicheng Ding, Min Li, Xianjie Han

TL;DR
This study analyzes the genomes of two Streptococcus parasuis strains from pigs in China to understand their potential zoonotic threat and drug resistance.
Contribution
The study identifies key virulence and antibiotic resistance genes in S. parasuis and links mobile genetic elements to drug resistance.
Findings
srtC, ctpV, and sugC are key virulence genes in S. parasuis, though their pathogenic potential is lower than in S. suis serotype 2.
S. parasuis shows resistance to aminoglycosides, macrolides, tetracyclines, and oxazolidinones but is susceptible to some oxazolidinone-class antibiotics.
Mobile genetic elements are associated with antibiotic resistance in the studied S. parasuis strains.
Abstract
(1) Background: S. parasuis is a potential opportunistic zoonotic pathogen that can infect pigs, cattle, and humans, composed of former members of S. suis serotypes 20, 22, and 26. In recent years, unclassified serotypes and a serotype 11 S. parasuis have been discovered. (2) Methods: We characterized two S. parasuis strains (FZ1 and FZ2) isolated from brain samples of paralyzed pigs and examined evolutionary divergence among 22 available S. parasuis and 8 serotype 2 S. suis genomes through whole-genome sequencing and comparative genomic analysis. We compared virulence genes (VGs) and antibiotic resistance genes (ARGs) and analyzed mobile genetic elements (MGEs) in FZ1 and FZ2. (3) Results: Comparative genomics revealed that srtC, ctpV, and sugC may represent key virulence determinants in S. parasuis, although their pathogenic potential appears attenuated compared to serotype 2 S. suis.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
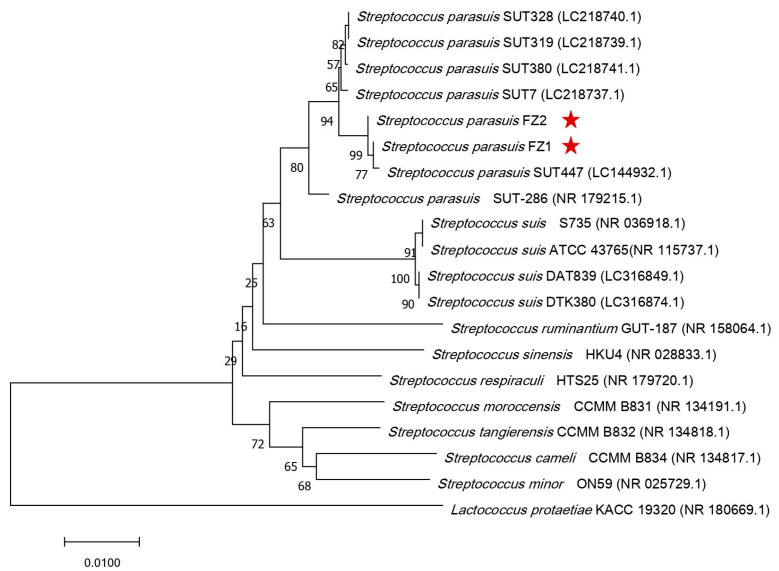 Figure 1
Figure 1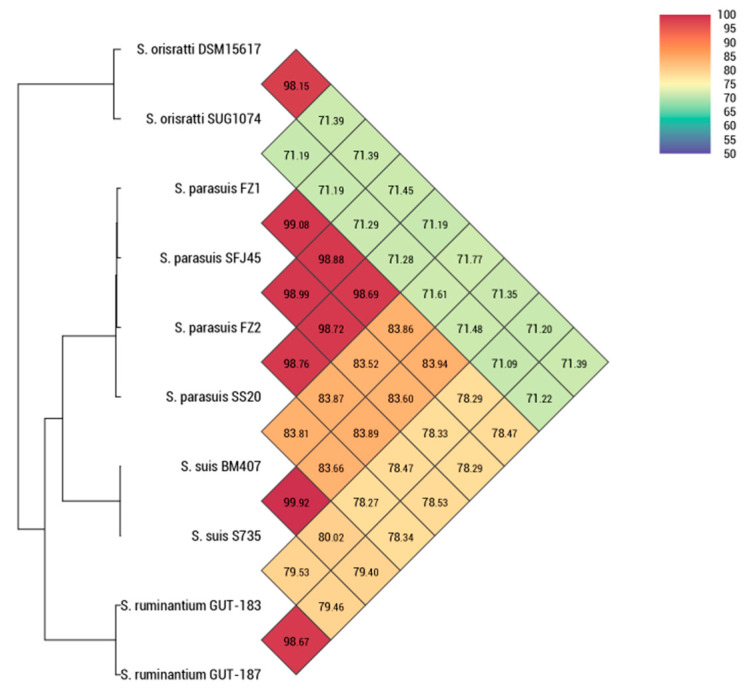 Figure 2
Figure 2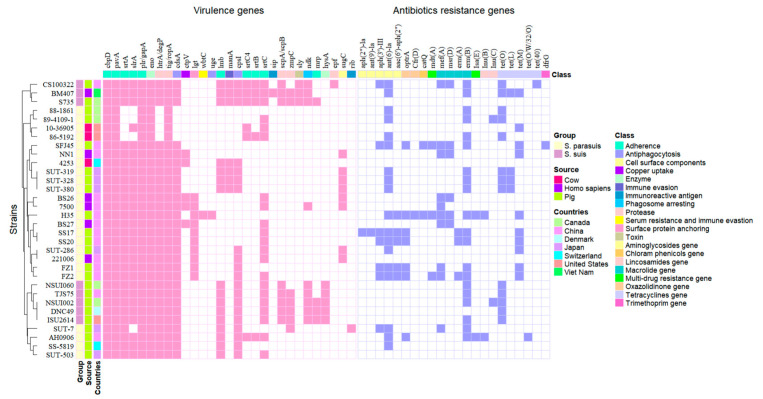 Figure 3
Figure 3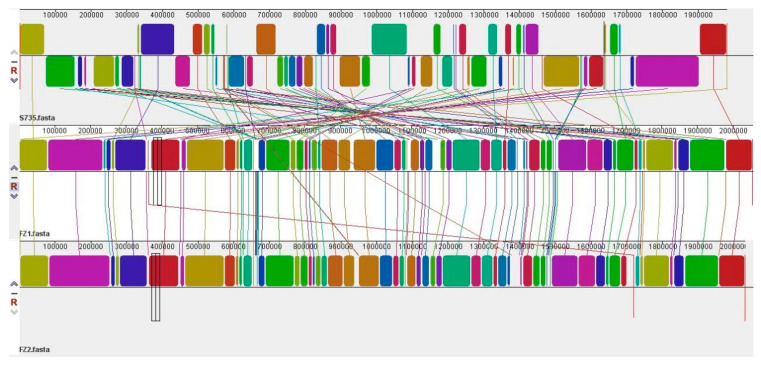 Figure 4
Figure 4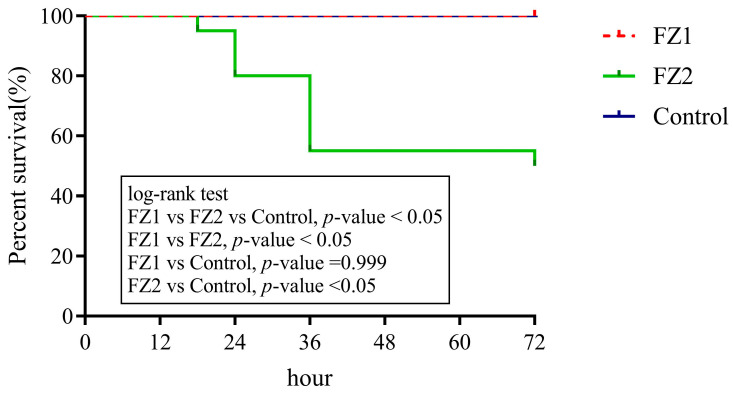 Figure 5
Figure 5- —National Natural Science Foundation of China
- —Natural Science Foundation of Shandong Province
- —Position for the Production and Environmental Control of Innovation Team in Pig Industry of Modern Agricultural Technology System in Shandong Province
- —Research Foundation for Distinguished Scholars of Qingdao Agricultural University
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsStreptococcal Infections and Treatments · Microbial infections and disease research · Rabies epidemiology and control
1. Introduction
Streptococcus suis (S. suis) is an important zoonotic pathogen that can be transmitted to humans via direct contact with infected pigs or consumption of undercooked contaminated pork products, constituting a potential risk to public health and safety [1]. S. suis is Gram-positive and can be divided into 35 serotypes according to different capsular polysaccharide antigens [1]. Serotype 2 S. suis is the most prevalent serotype with zoonotic potential, especially in Europe and Asia [2]. In 2015, S. suis serotypes 20, 22, and 26 were reclassified as Streptococcus parasuis (S. parasuis) on the basis of average nucleotide identity, 16S ribosomal RNA (rRNA), and biochemical characteristics [3]. In contrast to S. suis, clinical reports of S. parasuis remain limited, primarily due to the absence of reliable diagnostic methods capable of differentiating these species [4,5]. In 2018, a novel polymerase chain reaction (PCR) method specifically targeting S. parasuis was developed on the basis of nucleotide sequence comparisons of recN with S. parasuis and its close relatives [4].
The presence of S. parasuis in diseased pigs and calves with pneumonia or systemic infection (meningitis, arthritis, endocarditis, or septicemia) suggesting its pathogenic potential in livestock [6,7,8]. S. parasuis is widespread in swine populations globally, with only sporadic reports of infection in calves, and given the lack of clinical isolates, the importance of S. parasuis for public health is underestimated [3]. Recently, three human S. parasuis infection cases with pneumonia and arthritis have been reported in China, indicating that the zoonotic pathogen S. parasuis is an emerging threat to public health [3,9]. At present, the presence of S. parasuis has recently been reported in a few countries, such as China, Japan, Canada, and Switzerland [10], but the genome characteristics and pathogenesis of S. parasuis still need further study. In this study, two new strains of S. parasuis were isolated from the brains of two paralyzed pigs, the genomic characteristics of S. parasuis isolates FZ1 and FZ2 were analyzed, and evolutionary divergence was assessed through whole-genome sequencing and comparative genomic analysis of 22 S. parasuis genomes and 8 serotype 2 S. suis genomes. This study provides new insights into the genomic and evolutionary characteristics of S. parasuis and provides a new basis for the study of bacterial pathogenesis and drug resistance in the future.
2. Materials and Methods
2.1. Bacterial Isolation and Identification
In May 2023, brain samples were aseptically collected from two paralytic pigs at a pig farm in Shandong Province and promptly transported to the laboratory under refrigeration. Deep tissue samples were aseptically excised using sterile scissors and then incubated on blood agar plates at 37 °C for 24 h under aerobic conditions. Single colonies were selected and purified using blood agar, and the morphological characteristics of the isolated bacteria were determined by Gram staining and biochemical identification with the API 20 strep Streptococcus Biochemical Identification Kit (Biomerieux, Marcy I’Etoile, France). Two suspected strains of S. parasuis were named FZ1 and FZ2 and stored at −80 °C in broth containing 15% glycerol for further analysis.
2.2. Phylogenetic Analysis
Strains were grown aerobically in 25 mL of brain heart infusion broth (Haibo Biotechnology, Qingdao, China) at 37 °C with shaking at 180 rpm. For preliminary identification, a small fragment of the 16S rRNA gene was amplified using universal primer set (27F 5′-AGAGTTTGATCCTGGCTCAG-3′; 1492R 5′-GGTTACCTTGTTACGACTT-3′) [11]. The PCR mixture consisted of 8.5 μL of nuclease-free water, 12.5 μL of 2 × Taq Master Mix, 1 μL of each primer, and 2 μL of genomic DNA in a total volume of 25 μL. The PCR conditions were initial denaturation for 3 min at 95 °C, followed by 30 cycles of amplification for 15 s at 95 °C, 15 s at 55 °C, 90 s at 72 °C, and a final elongation at 72 °C for 5 min (GeneAmp PCR System 2700; Applied Biosystems, Foster City, CA, USA). PCR products were visualized by 1% (w/v) agarose gel electrophoresis in 1 × TAE buffer and then sequenced at Sangon Biotech Co., Ltd. (Shanghai, China). Spliced sequences were compared with online data in the NCBI database (http://www.ncbi.nlm.nih.gov, accessed on 5 April 2024), and multiple sequence alignments were carried out using the ClustalW program in MEGA 7.0 [12]. A phylogenetic tree was constructed by applying the neighbor-joining (NJ) method with the genetic distances calculated by the Kimura 2-parameter model. Bootstrap analysis was also performed in a total of 1000 replicates for the NJ analysis.
2.3. DNA Extraction
The genomic DNA was extracted following the instructions of the TIANamp Bacteria DNA Kit (TIANGEN BIOTECH, Beijing, China). Qualified genomic DNA was used as the starting material for sequencing and library construction. The quality and integrity of genomic DNA was assessed using 1% agarose gel electrophoresis and densitometry compared to the appropriate size standards. Meanwhile, DNA yield and purity were measured using NanoDrop™ 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and TBS-380 fluorometer (Turner BioSystems Inc., Sunnyvale, CA, USA). High-quality DNA was used to conduct further research.
2.4. Genome Sequencing, Assembly, and Annotation
In this study, a combination of the Illumina NovaSeq 6000 and PacBio Sequel II platforms was used to complete the genome maps of the two S. parasuis at Beijing Novogene Bioinformatics Technology Co., Ltd. (Beijing, China). An Illumina NovaSeq library and a PacBio library were constructed. Draft genome sequencing was carried out using the Illumina NovoSeq platform (Illumina, San Diego, CA, USA). Approximately 1 µg DNA was sheared to construct a sequencing library of 400~500 bp insertion fragments. Paired-end 2 × 150 bp reads were then sequenced. After quality control, the Illumina sequencing data were preliminarily assembled using SPAdes v3.15.3 [13]. For PacBio sequencing, SMRTbell library inserts (20 kb) were sequenced, and subreads shorter than 500 bp were removed. The PacBio sequences were error-corrected, binned, and then assembled through the Canu v2.2 assembler [14], and Pilon v1.24 [15] was used for assembly polishing with Illumina short reads to improve genome quality. To determine the presence of any plasmids, the filtered Illumina reads were mapped through SOAPdenovo2 [16] to the bacterial plasmid database. Circos v0.69-6 [17] was used to construct the circular map of the genome and comprehensively display the relevant information. The rRNA genes were identified through RNAmmer v1.2 [18], and the tRNA genes were identified through tRNAscan-SE v2.0.8 [19] with default settings. The above assembled sequences were used to predict coding genes through Glimmer3 v3.02. The predicted gene sequences were translated and searched against the National Center for Biotechnology Information (NCBI) nonredundant database, the Gene Ontology (GO) database, the protein families (Pfam) database, the Clusters of Orthologous Groups (COG) database, and the Kyoto Encyclopedia of Genes and Genomes (KEGG) database for annotation.
2.5. Comparative Genomic Analysis
To measure the similarity among the strains, the digital DNA-DNA hybridization (dDDH) and the Orthologous Average Nucleotide Identity (OrthoANI) were calculated between pairs of genomes. The dDDH was calculated with GGDC (Genome-to-Genome Distance Calculator 3.0 https://ggdc.dsmz.de/ggdc.php#, accessed on 3 May 2024). OrthoANI was calculated through ChunLab’s Orthologous Average Nucleotide Identity Tool (OAT), with an algorithm demarcation cutoff of 95~96% [20]. The genome sequences of FZ1 and FZ2 were submitted to PubMLST (https://pubmlst.org/, accessed on 10 May 2024) [21] and compared with the S. suis database.
Different online tools have been used to search for genetic transfer. The VFanalyzer tool of VFDB web-services (http://www.mgc.ac.cn/VFs/, accessed on 15 May 2024) [22] was used to estimate VGs. Amino acid sequences of VGs were compared with multiple-aligned sequences. ResFinder web-services (Center for Genomic Epidemiology (dtu.dk), accessed on 22 May 2024) [23] was used to detect ARGs and kept default settings. To search for CRISPR-Cas sequences, the genomes were analyzed through the CRISPRCasFinder (https://crisprcas.i2bc.paris-saclay.fr/, accessed on 23 May 2024) [24] online tool. PHASTEST (https://phastest.ca/, accessed on 25 May 2024) [25] was utilized for identifying prophage sequences; ICEfinder (https://tool2-mml.sjtu.edu.cn/ICEberg3/ICEfinder.php, accessed on 26 May 2024) [26] was used to detect ICEs. The identification of genomic islands was performed with IslandViewer 4 (https://www.pathogenomics.sfu.ca/islandviewer/, accessed on 26 May 2024) [27] through the IslandPath-DIMOB method.
In addition, all 22 sets of acquirable genome data for S. parasuis were downloaded from the National Centre for Biotechnology Information (NCBI) on June 2024, and pangenomic analysis of S. parasuis genomes including FZ1 and FZ2 was performed through BPGA v1.3 [28]. The raw sequencing dataset of S. parasuis SUT-319 was assembled through SPAdes v3.15.5 [13]. The VGs and ARGs of S. parasuis were compared with those of 8 serotype 2 S. suis strains that were randomly selected through R 4.3.2. A progressive Mauve (v2.4.0) [29] algorithm was introduced to observe the sequence identities of FZ1, FZ2, and S. suis S735. All S. parasuis and S. suis genomes used in this study are shown in Table 1.
2.6. Mice Survival Test
An experimental infection model in mice was designed to assess the pathogenicity of S. parasuis strains FZ1 and FZ2 by comparing the survival rates of infected mice. A total of 50 female C57BL/6 mice (6 weeks old) were randomly allocated into two infection groups (2 replicates per group, 10 mice per replicate) and one control group (2 replicates, 5 mice per replicate). C57BL/6 mice were intraperitoneally injected with 5 × 10^7^ CFUs of test strains in 1 mL of THB or THB alone (control). Mortality was recorded every 6 h for the first 24 h and every 12 h thereafter 72 h post-infection. Survival curves were generated using the Kaplan–Meier method, and experiments were performed in duplicate [9].
3. Results
3.1. Bacterial Characteristics and Phylogenetic Analysis of S. parasuis FZ1 and FZ2
The two isolated strains were demonstrated to be Gram-positive bacteria, and the colonies on blood agar were milky white, translucent, circular, and nonpigmented with α-hemolytic bacteria. Biochemical assays revealed that both strains tested negative for the pyruvate (VP) and hippurate (HIP) tests. Additionally, no enzymatic activity was detected for β-glucosidase (ESC), pyrrolidonyl arylamidase (PYRA), α-galactosidase (α GAL), β-glucuronidase (β GUR), β-galactosidase (β GAL), alkaline phosphatase (PAL), arginine dehydrolase (ADH), or leucine arylamidase (LAP). Acid was produced from trehalose (TRE), starch (AMD), and glycogen (GLYG), but not from ribose (RIB), L-arabinose (ARA), mannitol (MAN), sorbitol (SOR), lactose (LAC), inulin (INU), or raffinose (RAF).
To identify the pathogenic species accurately, a 1517 bp 16S rRNA sequence of the isolated strains was amplified and sequenced. BLASTN analysis confirmed that the sequences belonged to the genus Streptococcus. The 16S rRNA genes of strains FZ1 and FZ2 presented the greatest identification (>99%) with those of S. parasuis, with the highest sequence similarity with S. parasuis SUT-447 (99.93% and 99.86%, respectively), followed by those with S. parasuis SUT-7 (99.45% and 99.52%, respectively) and S. parasuis SUT-319 (99.43% and 99.50%). In contrast, the 16S rRNA of the isolates exhibited 96–97% identity with S. suis and 95–96% nucleotide identity with S. ruminantium.
To further verify the nucleotide BLAST results, a detailed phylogenetic tree was constructed, which revealed the relationships of S. parasuis FZ1 and FZ2 with closely related Streptococcus species. Moreover, the phylogenetic tree revealed that FZ1, FZ2, and S. parasuis SUT-447 clustered together in the same clade, demonstrating their high genetic relatedness. Distinct lineages were formed with S. parasuis SUT-7, S. parasuis SUT-380, S. parasuis SUT-328, S. parasuis SUT-319, and S. parasuis SUT-286 (Figure 1). Therefore, the phylogenetic analysis confirmed that the two isolated strains were novel strains of S. parasuis; thus, we designated them S. parasuis FZ1 and FZ2.
3.2. Genomic Features and Gene Functional Analysis of S. parasuis Isolates
The genomes of strains FZ1 and FZ2 consisted of 2,054,729 bp and 2,032,338 bp circular chromosomes with mean G+C contents of 39.46% and 39.44%, respectively. The complete genome sequences of FZ1 and FZ2 contained 2085 and 2125 predicted coding sequences (CDSs), respectively, while both contained 58 tRNAs, 12 rRNAs, 4 5S rRNAs, 4 16S rRNAs, and 4 23S rRNAs. In addition, three plasmids—pFZ2-1 (16,488 bp), pFZ2-2 (6065 bp), and pFZ2-3 (5488 bp) were identified in FZ2 (Figure S1).
The gene functions were predicted through GO, COG, and KEGG analyses. The COG-annotated genes of FZ1 and FZ2 were both divided into 23 COG subclasses, with 1697 and 1691 COG-annotated genes, respectively. The most enriched categories were “translation, ribosome structure, and biogenesis” (222 and 221 genes, respectively), followed by “amino acid transport and metabolism” (179 and 175 genes, respectively), “transcription” (140 and 132 genes, respectively), “replication, recombination, and repair” (110 and 121 genes, respectively), “carbohydrate transport and metabolism” (105 and 100 genes, respectively), and “cell wall/membrane/envelope biogenesis” (103 and 116 genes, respectively) (Figure S2).
GO analysis revealed that 560 and 551 protein-encoding genes in FZ1 and FZ2, respectively, were categorized into biological process, cellular component, and molecular function categories. The most annotated biological process function was translation (54 and 54 genes, respectively). The integral components of the membrane (119 and 109 genes, respectively) and cytoplasm (83 and 87 genes, respectively) were the top two enriched cellular components. For molecular functions, DNA binding (86 and 82 genes, respectively) and ATP binding (83 and 72 genes, respectively) were the most abundant (Figure S3).
KEGG analysis identified 1255 and 1228 annotated genes in FZ1 and FZ2, respectively, distributed across six categories. Among them, the most populated class was represented by metabolism pathways (929 and 926 genes, respectively), followed by genetic information processing (156 and 155 genes, respectively), environmental information processing (165 and 161 genes, respectively), human diseases (77 and 86 genes, respectively), cellular processes (71 and 68 genes, respectively), and organismal systems (28 and 28 genes, respectively). The most abundant KEGG pathways were the global and overview maps (365 and 368 genes, respectively) within the metabolism category, followed by amino acid metabolism (114 genes, entirely) and membrane transport (111 and 113 genes, respectively) (Figure S4).
3.3. Comparative Genomic Analysis of S. parasuis FZ1 and S. parasuis FZ2
The dDDH and OrthoANI values of the isolated strains S. parasuis FZ1 and FZ2 were compared with those of the S. parasuis, S. suis, S. orisratti, and S. ruminantium strains. The dDDH values of S. parasuis FZ1 and FZ2 compared with those of S. parasuis SFJ45 (92.10% and 90.70%, respectively), and S. parasuis SS20 (87.8% and 88.8%, respectively) exceeded the 70% cutoff points, whereas those of S. suis BM407 (28.6% and 28.30%, respectively), S. suis S735 (28.60% and 28.30%, respectively), S. ruminantium GUT-183 (23.60% and 23.40%, respectively), S. ruminantium GUT-187 (23.10% and 23.00%, respectively), S. orisratti SUG1074 (25.00% and 24.70%, respectively), and S. orisratti DSM15617 (27.20% and 26.70%, respectively) were all below the 70% cutoff points recommended for delineating species. Similarly, the OrthoANI values for FZ1 and FZ2 compared to S. parasuis strains exceeded the 95–96% cutoff, while those for S. suis, S. orisratti, and S. ruminantium were below this threshold, which indicated that the isolated strains S. parasuis FZ1 and FZ2 were S. parasuis (Figure 2).
S. parasuis were compared with the pubMLST database of S. suis. Seven allele sequences of aroA, cpn60, dpr, gki, mutS, recA, and thrA were found in both FZ1 and FZ2. Submission of allele profiles of seven housekeeping genes to the PubMLST database confirmed that FZ1 and FZ2 belong to two different sequence types; hence, following verification, two new sequence types (STs) were assigned to the respective allele combinations ST2909 and ST2910 (Table 2).
One type of CRISPR-Cas system (Type IC) was found in S. parasuis FZ1 but was absent in S. parasuis FZ2. The chromosome of FZ1 carried one phage, one ICE, and nine GIs, of which the ICE and two GIs carried AMR genes, while FZ2 carried three phages and eights GIs, and only one phage and one GI carried AMR genes (Table 3). These factors, along with others yet to be investigated, collectively determine the emergence and transfer of AMR between S. parasuis and different bacterial strains.
Pangenomic analysis was performed on FZ1, FZ2, and 22 available S. parasuis strains, which had a total of 28,368 core genes (1182 genes per genome), 15,794 accessory genes (average of 658.08 genes per genome), 1680 unique genes (average of 70 genes per genome), and 141 exclusively absent genes (average of 5.88 genes per genome). To investigate the phylogenetic relationships among these 24 S. parasuis isolates, a neighbor-joining tree was constructed on the basis of core genome alignment (Figure S5). The phylogenetic tree revealed distinct clustering patterns based on the isolation sources of the strains. S. parasuis isolated from humans (BS26, BS27, NN1, 7500, 221006) and cows (86-5192, 10-36905) formed two different clades, whereas S. parasuis isolated from pigs (SUT-7, 88-1861, SUT-328, SUT-319, SUT-380, SS-5819, FZ2, FZ1, SFJ45, H35, SS20, SS17, AH0906, SUT-286, 89-4109-1, and SUT-503) formed multiple clades (Figure S5). Moreover, S. parasuis from China (FZ1, FZ2, SFJ45, H35, SS17, and SS20) formed a large clade, with the exception of S. parasuis AH0906, whereas S. parasuis from other countries formed multiple clades (Figure S5). These findings indicate that the phylogenomic analysis of the core genome may reveal differences between strains according to their isolated sources and regions.
The proteins encoded by all the genes of S. parasuis were annotated in the Database of Clusters of Orthologous Genes (COGs). Only assigned COG functional genes were considered. The different functional preferences of the core, accessory, and unique were analyzed. Core genes were predominantly associated with COG categories J (translation, ribosomal structure, and biogenesis), E (amino acid transport and metabolism), and R (general function prediction only), which were present in a greater proportion compared to accessory and unique genes. In contrast, accessory genes of S. parasuis were more often associated with the COG categories R, K (transcription), L (replication, recombination, and repair), and G, and unique genes were more often associated with the COG categories M (cell wall/membrane/envelope biogenesis), K, L, G, and R (Figure S5). This finding indicates that the core genes of S. parasuis are preferred for different physiological and biological functions, and that the functions of accessory and unique genes are involved in genetic evolution, contribute to species diversity and provide selective advantages.
The 135 virulence genes of Streptococcus from VFDB were investigated in 24 S. parasuis and 8 S. suis strains, of which 29 virulence genes were present in these strains and were further analyzed (Figure 3). The analysis revealed that virulence traits related to adherence, enzymes, and proteases were detected consistently in all of the examined strains, such as adherence associated virulence genes cbpD (100.00%, 32/32), pavA (100.00%, 32/32), plr/gapA (100.00%, 32/32), the enzyme associated gene eno (100.00%, 32/32), and the protease gene tig/ropA (100%, 32/32). In addition, seven other virulence genes are associated with adherence, such as slrA (87.50%, 28/32), srtA (87.50%, 28/32), lmb (50%, 16/32), srtC4 (18.75%, 6/32), srtB (15.63%, 5/32), srtC (65.63%, 21/32), and mrp (12.50%, 4/32); three virulence genes are associated with antiphagocytosis, including cdsA (87.50%, 28/32), cpsI (62.5%, 20/32), and uge (3.13%, 1/32); hysA (15.63%, 5/32) is associated with enzymes; four genes, including htrA/degP (87.50%, 28/32), scpA/scpB (25.00%, 8/32), zmpC (21.88%, 7/32), and epf (3.13%, 1/32), are associated with proteases; and the other eight categories of virulence genes, including lgt (31.25%, 10/32), ndk (28.13%, 9/32), sugC (25.00%, 8/32), ctp V(15.63%, 5/32), manA (21.88%, 7/32), sly (9.38%, 3/32), wbtC (3.13%, 1/32), sip (3.13%, 1/32), and rib (3.13%, 1/32), were also identified.
Among the 29 virulence genes in S. parasuis, 75.86% (22/29) of the virulence genes were present in pig S. parasuis, 51.72% (15/29) present in human S. parasuis and 55.17% (16/29) present in cow S. parasuis. S. parasuis from pigs in Japan (58.62%, 17/29) and China (58.62%, 17/29) had the highest detection percentages, followed by S. parasuis from Switzerland (37.93%, 11/29) and Canada (20.69%, 6/29) from pigs. The percentages of lmb (100%, 8/8), cpsI (100%, 8/8), srtC (100%, 8/8), scpA/scpB (100%, 8/8), and ndk (100%, 8/8) detected in 8 S. suis type 2 strains were greater than those detected in 24 S. parasuis strains (33.33%, 50%, 54.17%, 0.00%, and 4.17%, respectively).
We found that the wbtC and uge genes of S. parasuis H35 were unique genes; the scpA/scpB, mrp, and hysA genes of SUT-319 were unique genes; and the zmpC and rib genes of SUT-7 were unique genes in S. parasuis. Moreover, the sip, scpA/scpB, epf, sly, mrp, and hysA genes are specific to S. suis, and the ctpV, lgt, wbtC, uge, sugC, and rib genes are specific to S. parasuis and exist only in human S. parasuis. These findings indicate that there are differences in the virulence genes of S. parasuis strains isolated from different sources and regions and that there are differences between S. parasuis and serotype 2 S. suis strains (Figure 3). In addition, compared with S. parasuis BS26, S. parasuis FZ1 lacks the srtC, ctpV, and sugC genes, whereas S. parasuis FZ2 lacks the ctpV and sugC genes. Combined with the results of the mouse survival test, we speculate that the srtC, ctpV, and sugC genes are important virulence factors for the pathogenicity of S. parasuis.
A total of 21 and 10 ARGs divided into eight categories were detected from S. parasuis and S. suis, respectively. The analysis revealed that ARGs related to aminoglycosides, macrolides, and tetracycline were detected consistently in S. parasuis and serotype 2 S. suis. The aminoglycoside gene ant(6)-Ia (62.50%, 15/24) and macrolide gene erm(B) (54.17%, 13/24) were the two genes with the highest detection percentages in S. parasuis, followed by macrolide mef(A) (37.50%, 9/24), tetracycline tet(M) (37.50%, 9/24), tet(O) (25.00%, 6/24), aminoglycoside aph(3′)-III (25.00%, 6/24), and oxazolidinone optrA (29.17%, 7/24). The aminoglycosides ARGs aph(2″)-Ia (4.17%), ant(9)-Ia (4.17%), and aac(6′)-aph(2″) (20.83%); the oxazolidinone ARG cfr(D) (4.17%); the chloram phenicols ARG cat(Q) (8.33%); the multidrug ARGs mdt(A) (12.50%) and Isa(E) (8.33%); the macrolide ARGs msr(D) (20.83%) and erm(A) (12.50%); the lincosamide ARGs lnu(B) (8.33%) and lnu(C) (4.17%); the tetracycline ARGs tet(L) (8.33%) and tet(O/W/32/O) (4.17%); and the trimethoprim ARG dfrG (4.17%) were also identified in 24 S. parasuis. In addition, the carriage of ARGs varies among different countries and source isolates. With respect to the isolated regions, Chinese S. parasuis from pigs had the richest resistance categories (8/8) of ARGs, followed by S. parasuis from pigs in Canada (4/8) and Japan (3/8). With respect to the isolated sources, S. parasuis from pigs contained the highest number of ARGs (21/21), followed by S. parasuis from cows (4/21) and humans (3/21). Moreover, erm(B) (75.00%, 6/8) and tet(O) (87.50%, 7/8) had the highest detection percentages in the studied strains of serotype 2 S. suis, followed by ant(6)-Ia (25.00%), aph(3′)-III (12.50%), mef(A) (12.50%), msr(D) (12.50%), tet(L) (12.50%), and tet(40) (12.50%), while the remaining ARGs were not detected in serotype 2 S. suis. These findings indicated that the antibiotic resistance of S. parasuis and S. suis was similar.
Collinearity revealed that the genomes of FZ1 and FZ2 presented obvious fragmented deletions, inversions, and translocations compared with those of S. suis S735, whereas the collinear set of matched colored regions of FZ1 and FZ2 presented significant similarities (Figure 4). These results suggest that although the two types of strains belong to Streptococcus, there are significant differences in their evolutionary histories. This may be related to their long-term independent evolution in different microenvironments. More in-depth analyses of the completely sequenced genomes of S. parasuis strains and investigations of the specific functions of important genes are necessary in the future.
3.4. Differential Survival Rates in Mice Infected with S. parasuis FZ1 and S. parasuis FZ2
Mice infected with S. parasuis FZ1 exhibited significantly higher survival rates compared to those challenged with FZ2 (p < 0.05). The survival rate of the mice infected with 5 × 10^7^ CFUs of S. parasuis strain FZ1 was 100% at 24 h post-infection, whereas that of the mice infected with strain FZ2 was 80% at the same time point. The survival rates of the mice infected with S. parasuis FZ1 and FZ2 were 100% and 50%, respectively, at 72 h post-infection (Figure 5).
4. Discussion
S. parasuis has been isolated from pigs, humans, and cows, and has characteristics similar to those of S. suis; however, its enzymatic activity and acid production differ [5]. S. parasuis is often isolated from healthy pigs, which has led to the notion that S. parasuis may be included in the normal microbiota of pigs. S. parasuis can be concomitantly isolated from diseased pigs and the bacterium itself has a low degree of virulence [4]. Exploring the genomic characteristics at the whole-genome level or comparing some phenotypic determinants could improve our understanding of the molecular and evolutionary changes in bacteria [9,30]. In this study, the genomes of S. parasuis isolates FZ1 and FZ2 were analyzed and compared with those of acquirable S. parasuis and its closely related serotype 2 S. suis genomes obtained from a public database.
To evaluate the potential virulence of S. parasuis strains FZ1 and FZ2, survival curves of FZ1 and FZ2 were generated, and virulence genes were compared with all available genomes of S. parasuis from public databases. Deficiency of the srtC gene significantly affects the synthesis of pili structure, leading to a significant reduction in bacterial adhesion, invasion, and virulence [31]. Mice infected with the ΔctpV strain (Mycobacterium tuberculosis) exhibit a reduced immune response to bacteria and a significantly increased lifetime [32]. The lifetime of mice infected with the ΔsugC mutant strain (Mycobacterium) through the aerosol pathway was significantly longer than that of those infected with the wild-type strain [33]. Compared with those in S. parasuis BS26, the ctpV and sugC genes in S. parasuis FZ1 and S. parasuis FZ2 were absent, and the srtC gene was absent in FZ1. In addition, significant differences in survival were detected between FZ1 and FZ2-infected groups and the BS26-infected group. Therefore, the srtC, ctpV, and sugC genes were crucial for the pathogenicity of S. parasuis, and their role in S. parasuis could be evaluated in further research.
The massive quantities of bacterial genomic data generated have facilitated in-depth analyses of bacteria for pangenomic studies [34]. Different numbers of accessory and unique genes were present in 24 S. parasuis. These dispensable genes and strain-specific genes are categorized as secondary genes, delineating the partially shared and strain-specific attributes of a species. These characteristics distinguish strains from one another and contribute to species diversity. Partially shared and strain-specific genes play roles that are not essential for growth but provide selective advantages, such as adaptation to different hosts and antibiotic resistance, indicating that owing to the presence of accessory genes and specific genes, S. parasuis is diverse. Structural variation in bacterial genomes is an important evolutionary driver. Genomic rearrangements, such as inversions, duplications, and insertions, can regulate gene expression and promote niche adaptation [35].
In addition, the distribution of virulence and antibiotic-resistant genes of S. parasuis in different strains may be correlated with the virulence of S. parasuis and the isolation source and regions of the strains. Genomic analysis revealed that S. parasuis and serotype 2 S. suis frequently harbor resistance genes for aminoglycosides, oxazolidinones, macrolides, and tetracyclines, indicating that antibiotic resistance is similar between S. parasuis and serotype 2 S. suis. Although oxazolidinones have not been approved for veterinary use, the potential for co-selection and co-transfer of oxazolidinone resistance genes may be facilitated by the widespread use of other antimicrobial agents, such as phenicols, in animal populations [36].
Various common ARGs and some high-risk ARGs (i.e., blaampC, blaOXA-1, and blaTEM-1) were prevalent in family livestock waste, and the pollution of tetracycline resistance genes was the most serious in these family livestock farms [37,38]. Infections caused by antibiotic-resistant bacteria are a major threat to global public health [39]. Mobile genetic elements (MGEs) play a key role in the intra- and interspecies horizontal transfer of antimicrobial resistance determinants. For example, several MGEs carrying ARG determinants for tetracyclines, macrolides, aminoglycosides, and chloramphenicol have been identified in S. suis [40]. Previously, a tet(M)- and aadE-carrying pathogenicity island (PAI) was identified in China, which was found to be unusual in streptococcal toxic shock syndrome (STSS)-causing S. suis strains from pigs. The tet(W)-carrying prophage ΦSsuD.1 was found in a S. suis strain from humans, and chimeric and tandem ICEs in streptococci have been reported [40]. In this study, different MGEs carrying ARGs were predicted in S. parasuis FZ1 and FZ2. Our findings indicate that there may be potential genetic exchange between the two strains and other bacterial strains, and that the interaction of MGEs in FZ1 and FZ2 may increase MGE diversity and complexity.
The collinearity analysis revealed structural variation in the evolutionary process of S. parasuis and serotype 2 S. suis. This may be related to their long-term independent evolution in different microenvironments. These analyses provide a basis for comparative genomics research and the study of genome-wide evolutionary dynamics.
5. Conclusions
In summary, this study sequenced the whole genomes of S. parasuis FZ1 and FZ2 strains in China and examined all the genomes of S. parasuis from public databases to explore their similarities and differences from those of its close relative serotype 2 S. suis. We clearly revealed that the srtC, ctpV, and sugC genes may be important for the pathogenicity of S. parasuis. There may be differences in virulence among different S. parasuis strains, which may be related to the isolation source and regions of the strains. In addition, S. parasuis is mainly resistant to aminoglycosides, macrolides, tetracyclines, and oxazolidinones, which is similar to serotype 2 S. suis, and there is an important association between MGEs and antibiotic resistance in S. parasuis FZ1 and FZ2. In addition, long-term independent evolution in different microenvironments may have led to structural variations between S. parauis and serotype 2 S. suis and differences in the virulence genes of the two types of Streptococcus. This study elucidates molecular mechanisms underlying antibiotic resistance and pathogenic virulence in S. parasuis, establishing a framework for future investigations into bacterial pathogenesis and antimicrobial resistance dynamics, and providing a better understanding of the evolution of S. suis.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Goyette-Desjardins G. Auger J.P. Xu J. Segura M. Gottschalk M. Streptococcus suis, an important pig pathogen and emerging zoonotic agent-an update on the worldwide distribution based on serotyping and sequence typing Emerg. Microbes Infect.20143 e 4510.1038/emi.2014.4526038745 PMC 4078792 · doi ↗ · pubmed ↗
- 2Guo G. Du D. Yu Y. Zhang Y. Qian Y. Zhang W. Pan-genome analysis of Streptococcus suis serotype 2 revealed genomic diversity among strains of different virulence Transbound. Emerg. Dis.20216863764710.1111/tbed.1372532654396 · doi ↗ · pubmed ↗
- 3Wang J.P. Yi X.L. Liang P.J. Tao Y.M.H. Wang Y. Jin D. Luo B. Yang J. Zheng H. Investigation of the Genomic and Pathogenic Features of the Potentially Zoonotic Pathogens 20211083410.3390/pathogens 1007083434357984 PMC 8308872 · doi ↗ · pubmed ↗
- 4Yamada R. Tien L.H.T. Arai S. Tohya M. Ishida-Kuroki K. Nomoto R. Kim H. Suzuki E. Osawa R. Watanabe T. Development of PCR for identifying Streptococcus parasuis, a close relative of Streptococcus suis J. Vet. Med. Sci.2018801101110710.1292/jvms.18-008329877313 PMC 6068303 · doi ↗ · pubmed ↗
- 5Nomoto R. Maruyama F. Ishida S. Tohya M. Sekizaki T. Osawa R. Reappraisal of the taxonomy of Streptococcus suis serotypes 20, 22 and 26: Streptococcus parasuis sp. nov Int. J. Syst. Evol. Microbiol.201565 Pt 243844310.1099/ijs.0.067116-025385995 · doi ↗ · pubmed ↗
- 6Wei Z. Li R. Zhang A. He H. Hua Y. Xia J. Cai X. Chen H. Jin M. Characterization of Streptococcus suis isolates from the diseased pigs in China between 2003 and 2007 Vet. Microbiol.200913719620110.1016/j.vetmic.2008.12.01519185432 · doi ↗ · pubmed ↗
- 7Gottschalk M. Lacouture S. Bonifait L. Roy D. Fittipaldi N. Grenier D. Characterization of Streptococcus suis isolates recovered between 2008 and 2011 from diseased pigs in Québec, Canada Vet. Microbiol.201316281982510.1016/j.vetmic.2012.10.02823177911 · doi ↗ · pubmed ↗
- 8Gottschalk M. Higgins R. Jacques M. Mittal K.R. Henrichsen J. Description of 14 new capsular types of Streptococcus suis J. Clin. Microbiol.1989272633263610.1128/jcm.27.12.2633-2636.19892480359 PMC 267098 · doi ↗ · pubmed ↗