Skeletal muscle effects of antisense oligonucleotides targeting glycogen synthase 1 in a mouse model of Pompe disease
Lan Weiss, Michele Carrer, Alyaa Shmara, Angela Martin, Hong Yin, Pallabi Pal, Cheng Cheng, Lac Ta, Victoria Boock, Yasamin Fazeli, Mindy Chang, Marvin Paguio, Jonathan Lee, Howard Yu, John Weiss, Tamar R Grossman, Nina Raben, Paymaan Jafar‐Nejad, Virginia Kimonis

TL;DR
Antisense oligonucleotides targeting glycogen synthase 1 reduce glycogen buildup in skeletal muscle and improve motor function in a mouse model of Pompe disease.
Contribution
Demonstrates ASO-mediated GYS1 knockdown as a novel therapeutic strategy to complement enzyme replacement therapy in Pompe disease.
Findings
ASO treatment significantly reduced glycogen accumulation in skeletal muscle of Gaa−/− mice.
Combining ASO with ERT improved muscle function and reduced autophagic buildup and lysosomal dysfunction.
Early administration of ASO with ERT may offer preventative treatment for Pompe disease.
Abstract
Pompe disease (PD) is a progressive myopathy caused by the aberrant accumulation of glycogen in skeletal and cardiac muscle resulting from the deficiency of the enzyme acid alpha‐glucosidase (GAA). Administration of recombinant human GAA as enzyme replacement therapy (ERT) works well in alleviating the cardiac manifestations of PD but loses sustained benefit in ameliorating the skeletal muscle pathology. The limited efficacy of ERT in skeletal muscle is partially attributable to its inability to curb the accumulation of new glycogen produced by the muscle enzyme glycogen synthase 1 (GYS1). Substrate reduction therapies aimed at knocking down GYS1 expression represent a promising avenue to improve Pompe myopathy. However, finding specific inhibitors for GYS1 is challenging given the presence of the highly homologous GYS2 in the liver. Antisense oligonucleotides (ASOs) are chemically…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
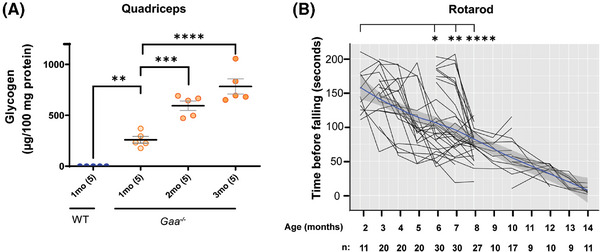 Figure 1
Figure 1 Figure 2
Figure 2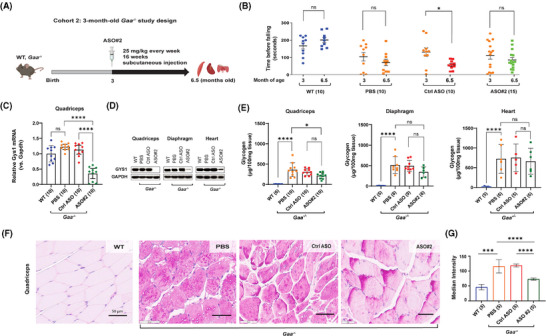 Figure 3
Figure 3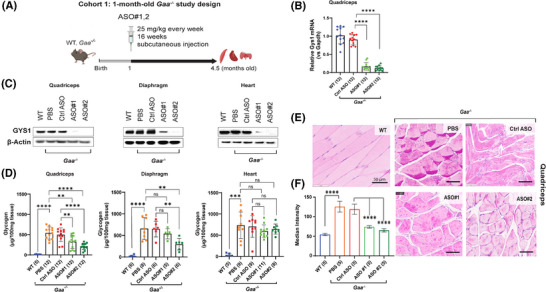 Figure 4
Figure 4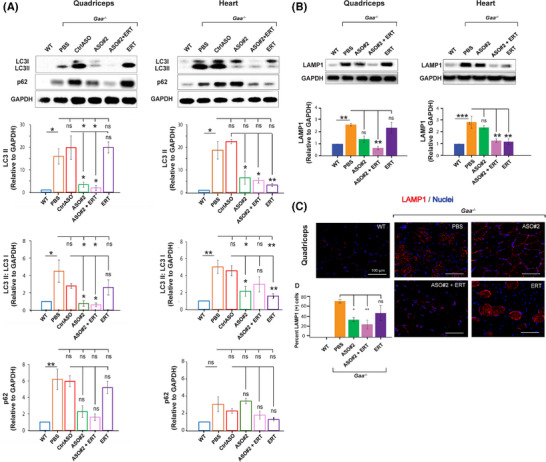 Figure 5
Figure 5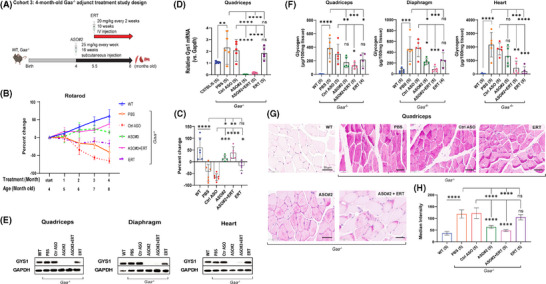 Figure 6
Figure 6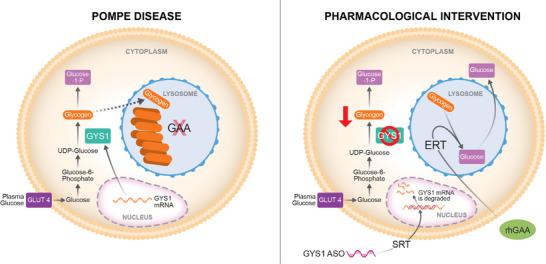 Figure 7
Figure 7Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsNeurogenetic and Muscular Disorders Research · Lysosomal Storage Disorders Research · Genetics and Neurodevelopmental Disorders