Checkpoint and recombination pathways independently suppress rates of spontaneous homology-directed chromosomal translocations in budding yeast
Li Zeng, Mingzeng Sun, Michael Fasullo

TL;DR
This study shows that checkpoint and recombination pathways in yeast independently prevent harmful chromosomal rearrangements caused by homologous recombination.
Contribution
The paper demonstrates that checkpoint and recombination pathways act independently to suppress homology-directed translocations in budding yeast.
Findings
Spontaneous recombination rates increased significantly in mutants defective in recombination or checkpoint pathways.
Double mutants lacking both checkpoint and recombination genes showed the highest increase in translocation rates.
Radiation-associated translocations also increased in mutants with defects in these pathways.
Abstract
Homologous recombination between short repeated sequences, such as Alu sequences, can generate pathogenic chromosomal rearrangements. We used budding yeast to measure homologous recombination between short repeated his3 sequences located on non-homologous chromosomes to identify pathways that suppress spontaneous and radiation-associated translocations. Previous published data demonstrated that genes that participate in RAD9-mediated G2 arrest, the S phase checkpoint, and recombinational repair of double-strand breaks (DSBs) suppressed ectopic recombination between small repeats. We determined whether these pathways are independent in suppressing recombination by measuring frequencies of spontaneous recombination in single and double mutants. In the wild-type diploid, the rate of spontaneous recombination was (3 ± 1.2) × 10−8. This rate was increased 10–30-fold in the rad51, rad55,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
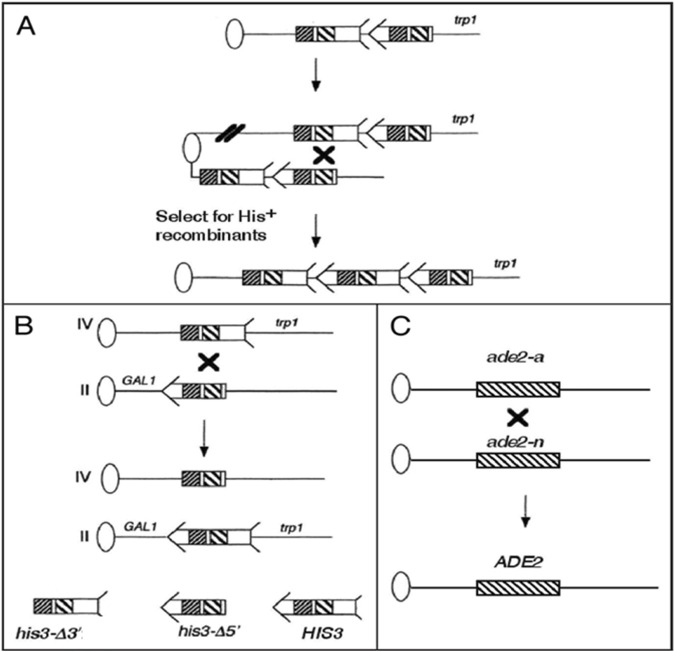 FIGURE 1
FIGURE 1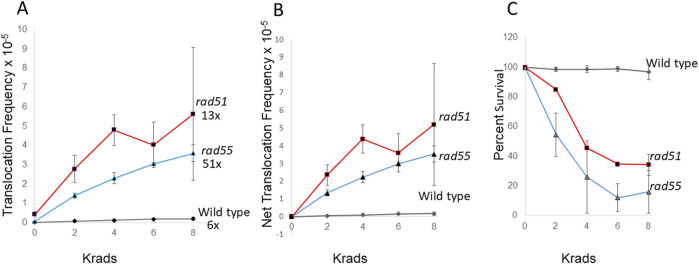 FIGURE 2
FIGURE 2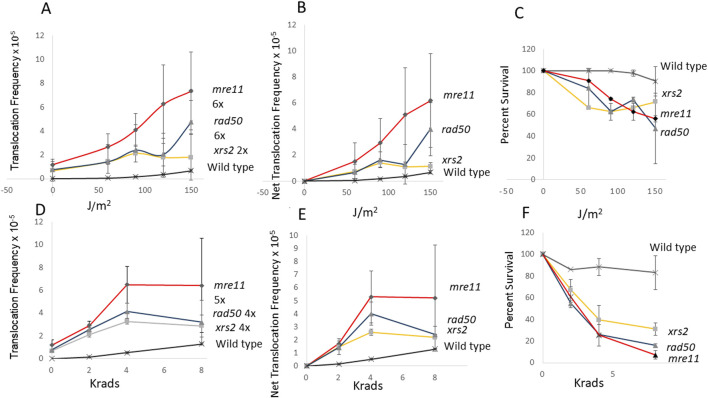 FIGURE 3
FIGURE 3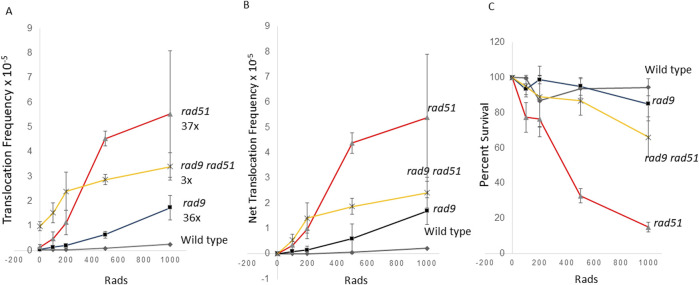 FIGURE 4
FIGURE 4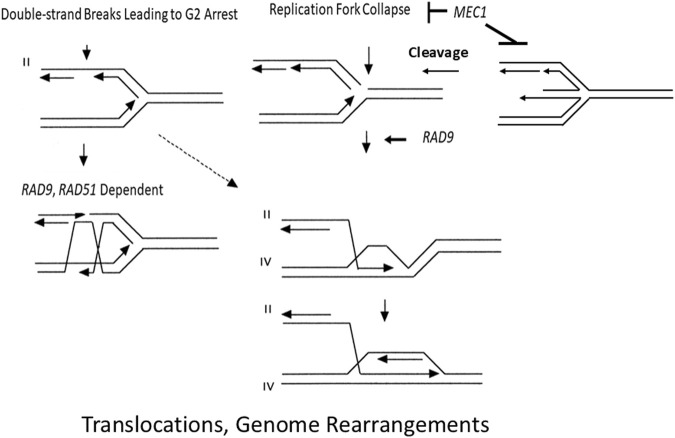 FIGURE 5
FIGURE 5| Relevant genotype | Number of experiments | Rate of translocations x 10−8
| Ratio |
|---|---|---|---|
| Wild type (YB110) | 5 | 3 ± 1.2 | 1 |
| Double-strand break repair | |||
|
| 3 | 34 ± 9 | 11 |
|
| 3 | 37 ± 6 | 12 |
|
| 3 | 38 ± 8 | 13 |
|
| 2 | 6.8 ± 0.1 | 2 |
|
| 3 | 67 ± 10 | 22 |
|
| 2 | 85 ± 7 | 28 |
|
| 2 | 80 ± 14 | 27 |
|
| 4 | 1.5 ± 0.4 | 0.5 |
|
| 2 | 7 ± 5 | 2 |
|
| |||
|
| 5 | 21 ± 5 | 7 |
|
| 2 | 180 ± 23 | 57 |
|
| 3 | 230 ± 62 | 77 |
|
| 3 | 203 ± 30 | 68 |
|
| 2 | 165 ± 50 | 55 |
|
| 2 | 130 ± 14 | 43 |
|
| 2 | 170 ± 57 | 57 |
|
| 2 | 58 ± 11 | 19 |
|
| 2 | 10 ± 0 | 3 |
| Genotype | Translocation (×10−8) | Ratio | Heteroallelic (×10−6)2 | Ratio | Sister Chromatid (×10−6) | Ratio |
|---|---|---|---|---|---|---|
|
| 3.0 ± 0.8 (YB348) | 1.0 | 0.9 ± 0.02 | 1.0 | 1.1 ± 0.1 (YB163) | 1.0 |
|
| 68 ± 16 (YB325) | 23 | 9.1 ± 1.9 | 10 | 6.3 ± 0.9 (YB311) | 5.7 |
|
| 33 ± 9 (YB742) | 11 | <0.01 | 0.01 | 2.3 ± 0.9 (YB177) | 2 |
|
| 291 ± 16 (YB757) | 100 | 1 ± 0.1 | 1.0 | 3.4 ± 1.9 (YB758) | 3.1 |
|
| 21 ± 5 (YB134) | 7 | 0.7 ± 0.2 | 0.8 | 1 ± 0.2 (YB147) | 1.0 |
|
| 19 ± 5(YB756) | 6 | 3.8 ± 1 | 4 | 1.8 ± 1(YB759) | 1.0 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsDNA Repair Mechanisms · Fungal and yeast genetics research · Carcinogens and Genotoxicity Assessment
Introduction
Homologous recombination between short repeated sequences can generate pathogenic chromosomal rearrangements; such recombination is often referred to as ectopic recombination. In mammalian cells, recombination between Alu sequences generates intrachromosomal deletions and translocations; several such rearrangements are associated with leukemias (Jeffs, et al., 1998; Strout et al., 1998; Balachandran et al., 2022). In budding yeast, recombination between delta sequences generates intrachromosomal deletions and inversions (Rothstein et al., 1987). While such events can spontaneously occur, they can also be stimulated by radiation and genotoxic agents, which lead to either replication fork collapse or double-strand breaks (DSBs, for review, see Nickoloff et al., 2023).
Eukaryotic cells have evolved genetic pathways to suppress ectopic recombination between repeats to minimize the occurrence of pathogenic rearrangements after exposure to environmental insults of DNA replication stress. These pathways include those that directly participate DNA repair and those that participate in cell-cycle checkpoint pathways which delay or arrest the cell cycle so that repair of DNA damage occurs before cellular division (Hartwell et al., 1994). Together these pathways assure that sister chromatids are the preferred substrates for recombinational repair of DSBs (Kadyk and Hartwell, 1992) and thus minimize ectopic recombination events that could generate rearrangements.
In budding yeast, DNA repair genes that function in DSB-repair by homologous recombination proteins constitute the RAD50 epistasis group (for review, see Krogh and Symington, 2004). Encoded proteins function to resect the ends of DSBs to generate recombinogenic 3′ single-strands, which in turn, are substrates for Rad51 filament formation, which catalyzes DNA strand invasion and the formation of Holliday intermediates. Additional proteins catalyze chromatin remodeling and resolve recombination intermediates. A recurrent theme is the presence of multiple Rad51 paralogs, nucleases, and resolvases. The identification of multiple mammalian orthologs and paralogs corresponding to yeast genes underscore the conservation of recombination functions in eukaryotic organism (Liu et al., 1998). Furthermore, defects in multiple mammalian recombination genes, such as BRCA1 and BRCA2, have been linked to cancer, underscoring the importance of elucidating the role of multiple recombination genes in maintaining genetic stability (for review, see Rein and Bernstein, 2023).
Checkpoint pathways have been divided into those that function at distinct stages in the cell cycle including the G_1_-S, S phase, and G_2_ phase. In budding yeast, the essential yeast gene, MEC1 (Kato and Ogawa, 1994), is the ataxia telangiectasia mutated and RAD3 related (ATR) ortholog, whose activation is facilitated by adaptors RAD9 and MRC1 (Sanchez et al., 1996). Downstream protein kinases are then required for the transcriptional DNA damage response, S phase delay, and the arrest of the cell cycle at the G_2_/M transition. MEC1’s signaling function in facilitating homologous recombination (Sanford et al., 2021; Xie et al., 2024) involves phosphorylation of key proteins, including Sgs1 (Hegnauer, et al., 2012), Rad51 (Flott et al., 2011) and Rad55 (Herzberg et al., 2006).
The role of genes in both the RAD50 epistasis group and checkpoint pathways in suppressing gross chromosomal rearrangements (GCRs) and intrachromosomal rearrangements has been well-documented. Importantly, knocking down both RAD51 and MEC1 confers a synergistic increase in GCRs (Pennaneach and Kolodner, 2004; Putnam and Kolodner, 2017), and DSBs are potent stimulators of GCRs (Myung and Kolodner, 2003). These studies underscore the importance of the S phase checkpoint in suppressing GCRs. However, these studies suggest that the checkpoint gene, RAD9, plays a minor role in suppressing GCRs (Myung et al., 2001).
Fasullo et al. (1998) illustrated the role of RAD9 in suppressing both spontaneous and DNA damage-associated homology-directed chromosomal translocations. These studies were performed in diploid and not haploid strains. The role of cell cycle arrest in suppressing radiation-associated chromosomal rearrangements was evident by observations that arresting rad9 mutants with the microtubule inhibitor nocodazole decreased elevated frequencies of radiation-associated translocations. Additional studies have shown the role of both RAD51 (Fasullo et al., 2004) and MEC1 (Fasullo et al., 2010) in suppressing homology-directed spontaneous and DNA damage-associated rearrangements. In these studies, electrophoretic karyotypes of recombinants revealed half-reciprocal translocations and additional rearrangements, suggesting that multiple recombination events occurred in mutant strains.
In this study, frequencies of homology-directed chromosomal rearrangements were measured in both single and double mutants defective in RAD9-mediated checkpoint function, S phase checkpoint function and recombinational repair. The overarching conclusion is that double mutants defective in both the recombinational DNA repair genes and either RAD9-mediated or S phase checkpoint function exhibit synergistic increases in rates of spontaneous recombination. Further experiments were performed to determine whether rad and checkpoint single and double mutants exhibit enhance radiation-associated recombination. We suggest that double mutants may be optimal strains for identifying effects of low dose radiation and the genotoxicity of environmental agents.
Methods
Growth media and transformations and yeast strains
Standard media were used for the culture of yeast and bacterial strains (Sherman et al., 1986). LB-AMP (Luria broth containing 100 μg/mL ampicillin) was used for the culture of the Escherichia coli DH1 strains. Media used for the culture of yeast cells included YPD (yeast extract, peptone, dextrose), SC (synthetic complete, 2% dextrose), SC-HIS (SC lacking histidine), and SC-ADE (SC-lacking adenine). YPD-Kan (G418) plates contain YPD supplemented with 50 μg/mL G418 (Sigma). YP(A)D contains YPD with 80 mg/L adenine. Gene knockouts were performed by one-step gene replacement (Rothstein, 1983), using standard lithium acetate transformation protocols (Gietz et al., 1995).
All the yeast strains, including BY4741 (Brachmann et al., 1998), and YA148 (Lesser and Guthrie, 1994) are of the S288c genetic background. The wild-type diploid strain containing the his3 recombination substrates (Fasullo and Davis, 1987) on one copy of chromosomes II and IV has been previously described. Checkpoint and recombination mutants were constructed by gene disruptions and genetic crosses. Essentially, two sets of isogenic haploid strains were constructed; one set contains the recombination substrates, and the other does not. The homozygous diploid strains, rad9 (YB134), rad51 (YB170), and mec1-21 (YB325), which also contain the recombination substrates, have been previously described (Fasullo et al., 1998; Fasullo et al., 2001; Fasullo et al., 2010). Additional homozygous diploid strains were obtained by knocking out RAD51, RAD55 RAD57, RAD50, RAD54, MRE11, XRS2 in respective MAT a and MATα haploid strains, using plasmids pRAD51Δ (Shinohara et al., 1992), pST11 (Lovett and Mortiner, 1987), pSM51 (Schild), pNKY83 (Alani et al., 1989), pSM31 (Schild), pSK-MRE11delta (Bressan et al., 1999), pET139 (Ivanov et al., 1994), respectively. The rad9 rad51, rad9 rad50, rad9 mre11, rad9 rad54 double mutants were constructed by knocking out the RAD genes in MATa rad9 and MATα rad9 haploids. The rad51 rad1 double mutants were obtained by knocking our RAD51 in rad1 haploid strains, and the diploid was obtained by mating the haploid double mutants. The rad51 rad9 double mutants were made by genetic crosses and mating the haploid double mutants. All disruptions were confirmed by radiation sensitivities and the haploid strains were then mated to generate the diploid strains used in this study. The radiation sensitivities of diploid mutants were confirmed by exposing inoculated YPD plates to 60 J/m^2^ and 6 krads of X rays and imaging after 3 days of 30°C incubation (see Supplementary Figure S1). PCR was used to confirm rad59::KanMX gene disruptions. The presence of the his3-Δ200 allele was confirmed by PCR analysis using the primers 5′-CACGGCAGAGACCAATCAGTA-3′ and 5′-GCACTCCTGATTCCGCTAATA-3’.
Haploids containing the recombination substrates for measuring unequal sister chromatid recombination (uSCR) and diploid strains containing ade2-a and ade2-n for measuring homolog recombination were previously described (Fasullo et al., 2010). The mec1-21 rad51 haploid strain containing the his3 recombination substrates was constructed by knocking out RAD51 in the mec1-21 haploids. Construction of the mec1-21 mutant diploid strains has been previously described (Fasullo et al., 2010). The mec1-21 rad9 double mutant was constructed by knocking out RAD9 in the MAT a and MATα mec1-21 haploids and then mating the haploid double mutants.
Determining the rates of spontaneous recombination
The rates of spontaneous, mitotic events that generate either SCE, heteroallelic or translocations were determined by the method of the median (Lea and Coulson, 1949), as performed by Esposito et al. (1982), using 11 independent colonies for each rate calculation. This method is essentially the same as that employed by Spell and Jinks-Roberston (2004). In all rate calculations, the number of cell divisions was estimated by the number of viable cells that formed colonies. Statistical analysis difference was determined by the by the Mann-Whitney U test, where there are at least two independent calculations for each mutant strain (Zar, 1996). To measure the rates of homolog recombination between ade2 heteroalleles, colonies were obtained from cells inoculated on YP(A)D solid medium to suppress expression of the adenine pathway. To determine whether the rate of recombination increased in the diploid mutants, compared to the single mutants, the interaction factor was calculated according to David et al. (2016), see Supplementary Table 3.
UV and X ray stimulation of recombination
The number of His^+^ recombinants stimulated by DNA-damaging agents was determined by subtracting the spontaneous frequency from the stimulated frequency and multiplying by 10^7^, the approximate number of cells plated, as done previously (Fasullo et al., 1994). The significance of the differences between mutants and Rad^+^ (diploid) strains was determined by using the two-tailed paired sample t-test (Zar, 1996). Protocols used to assess the recombinogenicity of UV and X rays have been described elsewhere (Fasullo et al., 1994). The X-ray radiation source was purchased from Rad Source, Inc. (Wheeling, Ill.), and the dose rate was 440 rads/min. For measuring radiation-associated stimulation of translocations, cells were washed twice in sterile H_2_O, resuspended in H_2_O, irradiated, and then plated on selective medium (SC-His) for selecting for recombinants and inoculating an aliquot of the appropriate dilution on YPD medium to measure viability. Typically, 20–200 colonies were counted on SC-HIS plates, and 100–300 colonies were counted on YPD plates. The UV source emitted 260 nm UV light at a dose rate of 2 J/m^2^. One-way ANOVA followed by Dunnet’s test was used to determine the statistical significance of differences between radiation-associated frequencies (Zar, 1996).
Results
RAD51, RAD51 paralogs or MRX genes suppress rates of spontaneous homology-directed translocations
Rates of spontaneous, mitotic translocations were measured in diploid and haploid strains using his3 constructs that are shown in Figure 1. We measured rates of spontaneous translocations in both diploid single and double mutants that were defective in RAD51, RAD55, RAD57 and the checkpoint gene RAD9. The rate of spontaneous translocations in the Rad^+^ diploid was 3 × 10^−8^ and was similar to previous measurements (4 × 10^−8^, average) obtained from three independent Rad^+^ diploids (Fasullo et al., 1998; Fasullo et al., 2004; Fasullo et al., 2010). Knocking out either RAD51, RAD55, or RAD57 conferred a ten-fold or greater increase in translocation frequency (Table 1). Diploid mutants defective in MRE11, *RAD50,*or XRS2, which encode the MRX complex, exhibited a 22–27-fold increase in the rate of homology-directed translocations. However, knocking out RAD54, only conferred a two-fold increase in recombination, compared to wild type, which was not significant (P > 0.05, Mann-Whitney), while knocking out RAD59, conferred a two-fold decrease; RAD54 functions at multiple stages in the initiation and processing of recombination intermediates (Heyer et al., 2006). These data indicate that knocking out the RAD51 paralogs, RAD55 and RAD57, confers a similar phenotype as that of knocking out RAD51, and that knocking out genes encoding the MRX complex conferred the highest increase in translocation frequencies among the RAD50 group. Thus, specific genes that function in recombinational repair of DSBs suppress homologous recombination between short repeated sequences on non-homologous chromosomes.
Sister chromatid, translocation, and heteroallelic recombination assays used in this study. Ovals represent centromeres and lines represent chromosomes. For simplicity, the left arms of the chromosomes are not included. An arrow and feathers together denote HIS3. As indicated in the bottom left of the figure, the 5′ deletion, his3-Δ5′, lacks the feather and the 3′ deletion, his3-Δ3′, lacks the arrow. The two regions of the sequence identity shared by the his3 fragments are indicated by decorated boxes; closely spaced diagonal-filled boxes indicate a region of 167 bp, and the broadly spaced diagonal line-filled boxes indicate a region of ∼300 bp. The “X” indicates where recombination occurred. (A) The his3-truncated fragments are integrated at the TRP1 locus to measure uSCR events. His+ recombinants resulting from unequal SCE were selected that contain HIS3 flanked by his3-Δ3′ and his3-Δ5'. (B) Homology-directed translocation events result from recombination between the same his3 fragments located each on chromosomes II and IV. Positions of the GAL1 and trp1 are shown on chromosomes II, IV, and the chromosomal translocations. (C) Homolog recombination between ade2-a and ade2-n generates ADE2. ADE2 and ade2 alleles are represented as boxes; ade2-a and ade2-n are separated by approximately 1 kb. Figure is an adaptation from Fasullo and Sun, (2008b).
Rates of homology-directed translocations are synergistically increased in mutants defective in both RAD9 and recombinational repair
Checkpoint and DNA repair mutants exhibit higher rates of spontaneous translocation events in diploid cells; however, it is unclear whether these pathways function independently to suppress recombination. We therefore determined whether knocking out both checkpoint and recombination genes would lead to a synergistic increase in recombination. Failure to arrest the cell cycle in G_2_ can also increase the frequency of DNA damage-associated chromosomal rearrangements. Compared to wild type, the rad9 diploid mutants exhibit a seven-fold increase in the rate of spontaneous recombination, similar to previous reports (Fasullo et al., 1998). We measured rates of translocations in double mutants defective in both rad9 and in recombinational repair genes (Table 1). The rate of translocations in rad9 mutants defective in either RAD51, RAD55, or RAD57 were similar and 57–77-fold higher than the wild-type rate. We also observed a 55-fold increase in rate of translocations in rad9 rad54 double mutant, compared to the rate in wild type. The fold increase in rates of the double mutant in comparison to the single mutants was thus synergistic, i.e., the increase was greater than additive (Perez-Perez et al., 2009), as indicated by the positive IF (see Supplementary Table 3). However, the synergistic increase in recombination is dependent on ploidy; the rate of spontaneous translocations was (2.0 ± 0.2) × 10^−8^ in the rad9 rad51 haploid, a rate below 5.4 × 10^−8^, observed in rad9 haploids. The higher rate of spontaneous recombination observed in rad51 diploids is also dependent on RAD59, indicating that RAD59 has a role in RAD51-independent ectopic recombination between these short-repeated sequences. This indicates that recombinational repair and RAD9 checkpoint and RAD51 participates in independent pathways for suppressing homology-directed translocation events in diploid strains.
The MRX complex is required in checkpoint signaling (Grenon et al., 2001). We also measured the rates of spontaneous recombination in mre11, xrs2, and rad50 diploid mutants that were defective in the RAD9 checkpoint function (Table 1). While rad9 mre11 and rad9 xrs2 double mutants exhibited over a 40-fold increase in rates of translocations., rates of translocations in rad50 and rad9 rad50 were still in the range of 20–25 ‐fold increase, compared to wild type. However, the radiation sensitivities of the rad9 mre11, rad9 xrs2, and rad9 rad50 mutants were uniformly higher than that of the mre11, xrs2, and rad50 single mutants (Supplementary Figure S1), and all the double mutants generate microcolonies after irradiation (Supplementary Table S2). Similarly, while there is a noticeable delay in the doubling time for the mre11, xrs2, and rad50 single mutants (100–110 min), the doubling time for the rad9 mre11, rad9 xrs2, and rad9 rad50 mutants was approximately the same as wild type (80–90 min). The data indicate that knocking out RAD9 in the mre11, xrs2 and rad50 single mutants increases radiation sensitivity while increasing the rate of translocations in either mre11 or xrs2 but not in rad50 mutants.
The MEC1-mediated S phase checkpoint and RAD51 independently suppress homology-directed translocations
Previous studies had shown that defects in S phase checkpoint function also increase translocations that result from homologous recombination between short repeats. The mec1-21 mutant (Craven and Petes, 2000) is a mec1 hypomorph that is defective in S phase checkpoint function but retains some G_2_ checkpoint function (Sun and Fasullo, 2007). We had previously measured recombination in the diploid mec1-21 mutant and observed a 23-fold increase in rates of spontaneous translocations, compared to the rate observed in the wild-type diploid (Fasullo et al., 2010). After RAD51 was knocked out in the mec1-21 diploid, the rate increased to 2.9 × 10^−6^, which is approximately 100-fold higher than what is observed in the wild-type diploid (Table 2). The rate of spontaneous translocations in the mec1-21 rad51 diploid is similar to the rate of spontaneous unequal siter chromatid exchange between juxtaposed his3 recombination substrates observed in Rad^+^ strains (Dong and Fasullo, 2003). However, knocking out RAD9 in the mec1-21 diploid decreased the rate of translocations, compared the rate observed in mec1-21, although it was still higher than wild type (Fasullo and Sun, 2008a). These data indicate that MEC1 and RAD51 participate in independent pathways for suppressing homology-directed chromosomal rearrangements. These data are consistent with the notion that genetic instability phenotypes are highest in S phase checkpoint mutants (Myung et al., 2001; Myung et al., 2004; Pennaneach and Kolodner, 2004).
We previously observed that the rates of spontaneous unequal sister chromatid recombination (uSCR) and homolog recombination between ade2 heteroallelic increased in mec1-21 mutants, compared to wild type (Table 2; Fasullo et al., 2010). While there is no significant difference in the rates of spontaneous uSCR in rad51 mutants and wild type, the rate of spontaneous, homolog recombination between ade2 heteroalleles cannot be detected (<1 × 10^−8^), in agreement with results obtained by Bai and Symington (1996). However, in diploid mec1-21 rad51 mutants recombination between ade2 heteroalleles was detected, and in the mec1-21 rad51 haploid the spontaneous rate of uSCR was three-fold higher than wild type. These data indicate that multiple types of homologous recombination events are enhanced in the mec1-21 strain, which are variably affected by knocking out RAD51.
Knocking-out RAD9 had no significant effect on the rates of unequal uSCR and homolog recombination between heteroalleles, consistent with observations that rad9 mutants do not generally exhibit mitotic phenotypes (Weinert and Hartwell, 1990). Similar to the RAD9 requirement for the high translocation frequencies observed in the mec1-21 diploid, knocking out RAD9 in diploid and haploid mec1-21 mutants also decreased homolog recombination and uSCR, respectively (Table 2). These data indicate diverse types of mitotic recombination events are increased in mec1-21 mutants which are variably affected by knocking out RAD51 and RAD9.
Both rad51 and rad55 diploid mutants exhibit enhanced radiation-associated recombination
The DNA lesions that initiate spontaneous translocation events are unknown. One explanation for the higher relative rates of spontaneous translocations in rad51 and rad55 diploid mutants is that spontaneously generated DSBs stimulate more homology-directed translocations in these rad mutants compared to those stimulated in wild type. In addition, both rad51 and rad55 mutants are defective in DNA damage-associated uSCR (Dong and Fasullo, 2003). We therefore exposed rad51 and rad55 diploid mutants to 2, 4, 6, and 8 krads of X rays and measured frequencies of X-ray associated translocations (Figure 2). For all exposures, the X-ray-associated frequencies of translocations in the rad mutants were significantly higher than wild-type frequencies (P < 0.05). We observed a dose-dependent increase in the frequencies of recombination wild type, rad51, and rad55 mutants (P < 0.05); however, the dose-dependent differences were most significant for the rad55 mutant (P < 0.01, Dunnett’s). Compared to the frequencies of spontaneous recombination, translocation frequencies after 8 krad exposure were 13-fold and 51-fold higher for rad51 and rad55, respectively, compared to 7-fold for wild type (Figure 2). The data indicate that frequencies of X-ray-associated translocations are similar in both the rad55 mutants and rad51 mutants and suggest that knocking-out other RAD51 paralogs may also confer increased X-ray associated genetic instability.
Stimulation of homology-directed translocations by ionizing radiation in wild type, rad51 and rad55 diploid strains. Radiation-associated recombination frequencies (translocations) were measured after rad51 (N = 3) and rad55 (N = 2), and wild type (N = 5) diploid strains were exposed to 0, 2, 4, 6, and 8 krads X rays. Recombination frequencies (A) were plotted against radiation dose. Net recombination frequencies were calculated by subtracting the spontaneous frequency from the radiation-associated frequency for each experiment (B). The average survival percentage was plotted against radiation dose (C). Shaded triangles represent the rad51 strain (YB170), shaded triangles represent the rad55 strain (YB744), and shaded diamonds represent the wild-type strain (YB110). Fold change was calculated by dividing 8 krad-associated frequency by the spontaneous frequency in (A).
mre11 diploid mutants exhibit more UV and X ray-associated recombination compared to wild type
Considering that mre11, rad50, and xrs2 diploid mutants also exhibit higher rates of spontaneous translocations, compared to wild-type diploid, we measured the radiation-associated translocations frequencies were higher in mre11, rad50, and xrs2 mutants (Figure 3). We exposed wild type, and mre11, rad50, and xrs2 mutants to both 2, 4, and 8 krads, as well as 60, 90, 120, and 150 J/m^2^. While the radiation-associated frequencies of translocations in wild type are significantly lower than all those in the rad mutants (P < 0.05), there is more than a 100-fold increase in UV-associated frequencies (6.8 × 10^−6^, avg.) and X-ray associated frequencies (1.3 × 10^−5^, avg.) in wild type, compared to the non-irradiated wild-type control (6 × 10^−8^, avg.). The frequencies of X-ray and UV-associated translocations were highest in the mre11 mutants and sixfold higher than the frequencies obtained from the non-irradiated control (Figure 3); the dose dependence is significant for both X-ray and UV exposures (P < 0.05, Dunnett’s). On the other hand, the frequencies of X-ray and UV-associated translocations are similar in the xrs2 and rad50 mutant, and while frequencies of UV and X-ray associated frequencies was dose-dependent for the xrs2 mutant (P < 0.05, Dunnett’s), only the X-ray associated frequencies were significant for the rad50 mutants (P < 0.01, Dunnett’s). These data illustrate that ectopic recombination between homologous sequences can be stimulated by radiation in mre11, rad50, and xrs2 mutants, which are defective in the processing of DSBs.
Stimulation of homology-directed translocations by UV and ionizing radiation in wild-type, mre11, rad50, and xrs2 diploid strains. In panel (A), UV-associated recombination frequencies (translocations) were measured after wild type (N = 2), mre11 (N = 4), rad50 (N = 2), and xrs2 (N = 3) diploid strains were exposed to 60, 90, 120 and 150 J/m2 UV. Net recombination frequencies were calculated by subtracting the spontaneous frequency from the radiation-associated frequency for each experiment (B). The average survival percentage was plotted against radiation dose (C). X-ray associated recombination were measured after wild type (N = 2), mre11 (N = 4), rad50 (N = 2), and xrs2 (N = 3) diploid strains were exposed 0, 2, 4, and 8 krads X rays (Panel D). Recombination frequencies (D) were plotted against radiation dose. Net recombination frequencies were calculated by subtracting the spontaneous frequency from the radiation-associated frequency for each experiment (E). The average survival percentage was plotted against radiation dose (F). Filled diamond represents the mre11 strain (YB743), filled triangle represents the rad50 strain (YB746), and the filled box represents the xrs2 strain (YB747), and the star represents the wild-type strain. (YB348). Fold change was calculated by dividing radiation-associated frequency by the spontaneous frequency in (A, D).
Radiation exposures less than 1 krad can increase translocation frequencies in rad9, rad51, and rad51rad9 diploids
We previously observed that the frequencies of radiation-associated rearrangements increase in rad51 (Fasullo et al., 2004) and rad9 mutants (Fasullo et al., 1998), defective in homologous recombination and G_2_ checkpoint control, respectively. However, it was unclear whether radiation-associated translocations could be detected at radiation exposures less than 1 krad (10 Gy); on average, 0.58 DSBs are introduced per krad in a diploid yeast cell (Frankenberg-Schwager and Frankenberg, 1990; Westmoreland and Resnick, 2016). We therefore determined whether rad9, rad51, and rad9 rad51 diploid mutants exhibit higher levels of radiation-associated translocations frequencies after exposures of less than 1 krad. We measured translocations frequencies after exposure to 1 krad, 500 rads, 200 rads and 100 rads of X-radiation (Figure 4). While after 1 krad X-ray exposure we observed a seven-fold increase in the frequencies of translocations in wild type diploids, we observed a 36-fold and 47-fold increase in radiation-associated frequencies in rad9 and rad51 mutants, respectively, but only a three-fold increase in the frequencies of translocations in the double rad9 rad51 double mutant. However, in the Rad^+^ diploid we did not observe higher levels radiation-associated frequences after cells were exposed to lower levels of radiation (Figure 4). Collectively, frequencies of radiation-associated translocations were significantly different in rad9, rad51, rad9 rad51 after radiation exposures at all doses, compared to frequencies of spontaneous translocations (ANOVA, P < 0.05); radiation-associated frequencies in rad9, rad51, and rad9 rad51 mutants after exposure to 500 rad were all significantly different than frequencies of spontaneous translocation. rad9 rad51 mutant showed the highest dose-associated significance after 200 rad (Dunnett’s, P < 0.05), 500 rad (Dunnett’s, P < 0.01, and 1 krad (Dunnett’s, P < 0.001), compared to the other strains. Compared to net frequencies of translocations after 100 rad exposures in wild-type cells, those obtained in rad51 (Dunnett’s, P < 0.04) and in rad51 rad9 cells (Dunnett’s, P < 0.003) were significantly different. Interestingly, the rad9 rad51 diploid mutant was more resistant to ionizing radiation at low radiation doses than the rad51 mutant. We speculate that the rad9 rad51 diploid mutant may be able to tolerate low levels of radiation than rad51 mutant due to lack of checkpoint function. On the other hand, we speculate that His^+^ recombinants generated in rad9 rad51 mutants after exposure to higher levels of radiation may contain more chromosomal rearrangements or chromosomal loss events, which may limit their viability. These studies thus indicate that frequencies of radiation-associated rearrangements increase in yeast mutants defective in DSB repair at exposures to low doses (100 rads) of ionizing radiation.
Stimulation of homology-directed translocations by “low dose” ionizing radiation in wild-type, rad9, rad51, and rad9 rad51 diploid strains. Radiation-associated recombination frequencies (translocations) were measured after wild type, rad51 (N = 3), rad9 (N = 3) and rad9 rad51 (N = 3) diploid strains were exposed to 100, 200, 500, 1,000 rads X rays. Recombination frequencies (A) were plotted against radiation dose. Net recombination frequencies were calculated by subtracting the spontaneous frequency from the radiation-associated frequency for each experiment (B). The average survival percentage was plotted against radiation dose (C). Filled triangle is the rad51 strain (YB170), filled box is the rad9 strain (YB134), filled diamond is the wild-type strain (YB110), and star is rad9 rad51 (YB749). Fold change was calculated by dividing 1 krad-associated frequency by the spontaneous frequency in (A).
Discussion
DNA repair and cell cycle checkpoint mutants exhibit higher frequencies of spontaneous chromosomal rearrangements. While higher frequencies of GCRs have been well-documented in rad and checkpoint yeast mutants (Myung et al., 2004; Smith et al., 2004), fewer studies have been performed regarding spontaneous and DNA damage-associated translocations generated by recombination between small repeat sequences, similar in size to human Alu sequences. We previously documented that higher frequencies of spontaneous and DNA damage associated translocations occur in rad51 (Fasullo et al., 2001), rad9 checkpoint mutants (Fasullo et al., 1998), and in mec1-21 hypomorphic mutants defective in the S phase checkpoint (Fasullo et al., 2010). In this study, we determined whether the RAD51-mediated recombination pathway, the RAD9-mediated checkpoint pathway, and the S phase checkpoint pathway are independent pathways in suppressing homology directed translocations. Our overall conclusion is that knocking out functions in both the RAD9 checkpoint and DSB repair pathways confers a synergistic increase in the rates of spontaneous translocations, while knocking down MEC1-mediated S phase checkpoint function and DSB repair conferred the highest frequencies of spontaneous translocations. In turn, specific rad mutants exhibit higher frequencies of radiation-associated translocations, even at relative low X-ray radiation exposures (100 rads). These studies are the first studies to document that radiation can stimulate homology-directed translocations in mutants defective in the MRX complex. We discuss our conclusions in the context of ploidy, comparisons with other studies performed in yeast, and implications for future studies in mammalian organisms.
We based our conclusions on results obtained by measuring translocation frequencies in (a/α) diploid mutants containing his3 recombinational substrates positioned on chromosomes II and IV, containing 300 bp of continuous sequence similarity. The advantage to measuring homology-directed translocations in diploid strains is that MAT heterozygosity suppresses non-homologous end joining by repressing NEJI (Kegel et al., 2001) and enhances DSB repair by homologous recombination (Fasullo and Dave, 1994). In addition, diploid His^+^ recombinants containing either non-reciprocal or reciprocal translocations are viable; haploids containing non-reciprocal translocations would not be viable due to the elimination of essential genes. However, frequencies of His^+^ recombinants generated by recombination between his3 truncated fragments only measure a subset of possible outcomes of recombination between short repeated sequences; for example, gene conversion events are not detected. Previous characterization of electrophoretic karyotypes exhibited by rad9 (Fasullo et al., 1998) and rad51 mutants (Fasullo et al., 2001) suggest that non-reciprocal and additional chromosomal rearrangements besides the directed translocations are also present and are currently under investigation.
Our results may initially seem to contradict results indicating that yeast ploidy decreases genetic instability (Tourrette et al., 2007). This study used a ura2 15-30-72 mutated allele construct in which Ura^+^ revertants contain complex rearrangements or mutations. Although the frequency of Ura^+^ revertants are decreased in diploid strains, there is an enrichment of Ura^+^ revertants that contain non-reciprocal translocations. Thus, while the frequencies of specific types of genetic instabilities may decrease with an increase in ploidy, it may also increase frequencies of genome rearrangements that would potentially confer lethality in haploids and are thus consistent with our studies. Since mammalian cells are diploid and cancer cells can be polyploid, diploid cells may represent a better understanding of genetic instability phenotypes (Putnam and Kolodner, 2017).
Our studies support the idea that RAD52 epistasis genes and checkpoint genes are independent pathways that ensure that sister chromatids are preferred templates for DSB repair, and when these functions are defective, there is a higher frequency that repeated sequences present on non-homologous chromosomes are utilized for recombinational repair. An attractive notion is that while RAD9 may delay the cell cycle to allow sufficient time for sister chromatid recombination (Fasullo et al., 1998; Mathiasen and Lisby, 2014), RAD51 promotes damage-induced sister chromatid recombination and suppresses alternative recombination pathways that generate half-reciprocal or reciprocal translocations. Thus, the synergistic increase in translocation frequency observed in rad9 rad51 double mutants may result from the persistence and accumulation of chromosomal fragments and the elevation of alternative pathways for repair of DSBs, such as single-strand annealing (SSA). While the precise mechanisms that generate non-reciprocal and reciprocal translocations in rad51 and rad9 mutants are unknown, translocations can be generated by break-induced replication (BIR, Sakofsky and Malkova, 2017) and SSA (Manthey and Bailis, 2010). BIR can also occur in rad51 mutants but requires RAD50 (Signon et al., 2001). SSA is an attractive mechanism since it is suppressed by RAD51 (Gallagher et al., 2020) and requires RAD59 (Pannunzio et al., 2012), which was required for the elevated rate of translocations observed in the rad51 diploid (Table 1). Deficient inhibition of Sgs1 in rad9 mutants (Ferrari et al., 2020) and de novo telomere formation (Marcomini et al., 2018) could also result in non-reciprocal chromosomal rearrangements. Thus, further characterization of chromosomal rearrangements in rad9 rad51 mutants is required to elucidate possible mechanisms.
Our surprising result was that mre11, rad50, xrs2 mutants higher frequencies of radiation-associated translocations and that rad9 mre11 and rad9 rad50 mutants exhibited synergistic increases in rate of spontaneous translocation, considering that MRX genes are required for the processing of DSBs and precede RAD9 in the checkpoint response to DSBs (Usui et al., 2001). Previous studies have indicated that redundant nucleases may resect the ends of DSBs (for reviews see, Mimitou and Symington, 2009; Cejka and Symington, 2021), among which are Mre11, Exo1 and Dna2, in cooperation with the Sgs1 helicase (Zhu et al., 2008). Our studies thus provoke questions as to which nucleases function in generating the recombinogenic 3′ ends that are required for homology-directed translocations. However, Westmoreland and Resnick (2016) demonstrate that radiation-associated cross-over events occur in mutants that exhibit little resection and Ivanov et al. (1994), observed that mating type switching occurs in xrs2 and rad50 mutants. In addition, Marcomini et al. (2018) observed that MRX deficiencies lead to greater mobility of chromosomal fragments, which could facilitate the formation of chromosomal rearrangements. Our data thus underscore that MRX complex suppresses ectopic recombination between repeated sequences (Oh and Symington, 2018).
The higher rates of spontaneous translocations in MRX mutants are consistent with studies that have shown that both rad50 (Malone et al., 1990) and mre11 (Bressan et al., 1999) mutants exhibit higher rates of heteroallelic recombination and cross-overs. Interestingly, rad50 rad9 double mutants did not exhibit enhanced rates of spontaneous translocation, compared to rates observed in the rad50 mutant, while mre11 rad9 and xrs2 rad9 double mutants do exhibit enhanced rates, compared to the single mutants (Table 1). Although the exact reason for differences in recombination phenotypes are unknown, previous studies have shown that mms2 rad50 and mms2 xrs2 double mutants exhibit different UV sensitivities, compared to the single mutants (Gangavarapu et al., 2007). In addition, Hailemariam et al. (2019), demonstrated that RAD50 but not MRE11or XRS2 was absolutely required for TEL1 activation. Thus the genetic instability phenotypes of rad50 may be different than that of xrs2 or mre11.
In agreement with the genetic control of GCRs (Pennaneach and Kolodner, 2004), our results indicate that the S phase checkpoint and RAD51 participate in independent pathways for suppressing chromosomal rearrangements (Figure 5). We speculate that lesions that initiate homology-directed translocations in mec1 mutants are generated by DNA replication fork collapse and that rad51 mutants are defective in replication fork maintenance and replication restart. Our data indicate that such lesions also stimulate recombination between sister chromatids and homologs (Table 2). RAD51 may also function to maintain DNA replication forks by non-recombination mechanisms, such as translesion DNA synthesis (Cano-Linares et al., 2021). However, in contrast to genetic control of GCRs, we previously observed that the highest frequencies of rearrangements occur in the mec1-21 mutant, which retains some G_2_ checkpoint function, and not the mec1 null mutant, which is defective in both S phase and G_2_ checkpoint function (Fasullo and Sun, 2008b). In our studies, the mec1-21 rad9 double mutant exhibits lower rates of translocations, compared to mec-21 (Fasullo and Sun, 2008a). Thus, we suggest that RAD9-mediated cell cycle delay is required to some of the recombination events that are initiated when replication fork collapse (Figure 5).
A proposed model of how defective gap repair in rad51 and rad9 mutants could enhance ectopic recombination between his3 fragments, resulting in the generation of a nonreciprocal translocation involving chromosomes II and IV. Each line represents a single strand of DNA, and the arrow indicates the 3′end. Chromosomes II and IV are designated by roman numerals. (Left) The DSB occurs at a site on the sister chromatid after polymerase progression and initiates SCE by gap repair. RAD51 and RAD9 function in redundant pathways to promote recombinational repair and suppress ectopic recombination. (Right) The DSB occurs because of a collapsed replication fork are after replication fork regression. Replication is then reinitiated after the 3′ end of the broken chromatid invades the intact sister chromatid. BIR can generate non-reciprocal translocations if the 3′ end of the broken chromatid invades an intact nonhomologous chromosome. The dashed arrow indicates the possibility that a DSB, which cannot be repaired by gap repair in a rad51 mutant, can initiate BIR and generate a nonreciprocal translocation. DSBs occurring in S phase require RAD9 function to efficiently initiate repair of collapsed forks. The figure was adapted from Fasullo et al. (2010).
The additional checkpoint mutants we constructed are particularly useful in detecting the recombinogenic effects of low dose exposure to genotoxic agents, as previously observed for the rad9 mutant (Fasullo et al., 2004). For example, relatively low doses of X rays (100 cGy) can stimulate chromosomal rearrangements in the rad9 rad51 diploid mutant. This dose generates less than one DSB per diploid genome but would be sufficient to generate at least one single-strand break (Roots et al., 1990). The threshold dose for detecting radiation effects in budding yeast has yet to be determined. We suggest that checkpoint mutants containing the recombination substrates would also be useful in detecting genotoxic damage by other agents, such as topoisomerase inhibitors (Fasullo et al., 2004), which are known to cause replication fork collapse or DSBs.
Our observations have relevance to Alu recombination in mammalian cells. DSBs targeted to Alu sequences have been shown to initiate translocations by SSA mechanisms (Elliott et al., 2005). The observation that knocking out RAD51 paralogs also confers genetic instability in yeast raises the question of whether deficiencies in other yeast or mammalian Rad51 paralogs (Liu et al., 1998) also confers higher frequencies of homology-directed rearrangements (Morales et al., 2021). Such studies may elucidate the role of polymorphisms in human X-ray repair genes (XRCC) associated with cancer (Liu et al., 2023).
In conclusion, we identified genes in independent pathways that suppress homology-directed translocations. These pathways underscore the role of the RAD9-mediated checkpoint pathway in suppressing homology-directed translocations and in enhancing recombination when the S phase checkpoint fail. It will be interesting to identify whether similar independent pathways suppress recombination between human Alu repeats.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Alani E.Subbiah S.Kleckner N. (1989). The yeast RAD 50 gene encodes a predicted 153-k D protein containing a purine nucleotide-binding domain and two large heptad-repeat regions. Genetics 122, 47–57. 10.1093/genetics/122.1.47 2659437 PMC 1203691 · doi ↗ · pubmed ↗
- 2Bai Y.Symington L. S. (1996). A Rad 52 homolog is required for RAD 51-independent mitotic recombination in Saccharomyces cerevisiae . Genes Dev. 10, 2025–2037. 10.1101/gad.10.16.2025 8769646 · doi ↗ · pubmed ↗
- 3Balachandran P.Walawalkar I. A.Flores J. I.Dayton J. N.Audano P. A.Beck C. R. (2022). Transposable element-mediated rearrangements are prevalent in human genomes. Nat. Commun. 13, 7115. 10.1038/s 41467-022-34810-8 36402840 PMC 9675761 · doi ↗ · pubmed ↗
- 4Brachmann C. B.Davies A.Cost G. J.Caputo E.Li J.Hieter P. (1998). Designer deletion strains derived from Saccharomyces cerevisiae S 288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14, 115–132. 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA 204>3.0.CO;2-2 9483801 · doi ↗ · pubmed ↗
- 5Bressan D. A.Baxter B. K.Petrini J. H. (1999). The Mre 11-Rad 50-Xrs 2 protein complex facilitates homologous recombination-based double-strand break repair in Saccharomyces cerevisiae . Mol. Cell. Biol. 19, 7681–7687. 10.1128/MCB.19.11.7681 10523656 PMC 84807 · doi ↗ · pubmed ↗
- 6Cano-Linares M. I.Yáñez-Vilches A.García-Rodríguez N.Barrientos-Moreno M.González-Prieto R.San-Segundo P. (2021). Non-recombinogenic roles for Rad 52 in translesion synthesis during DNA damage tolerance. EMBO Rep. 22 (1), e 50410. 10.15252/embr.202050410 33289333 PMC 7788459 · doi ↗ · pubmed ↗
- 7Cejka P.Symington L. S. (2021). DNA end resection: mechanism and control. Annu. Rev. Genet. 55, 285–307. 10.1146/annurev-genet-071719-020312 34813349 · doi ↗ · pubmed ↗
- 8Craven R. J.Petes T. D. (2000). Involvement of the checkpoint protein Mec 1p in silencing of gene expression at telomeres in Saccharomyces cerevisiae . Mol. Cell Biol. 20, 2378–2384. 10.1128/MCB.20.7.2378-2384.2000 10713162 PMC 85413 · doi ↗ · pubmed ↗