Attempts toward a Silyl-Stabilized Dicoordinate Borylene: Insertion of Carbon Dioxide into the B–Si Bond
Kasperi M. Salonen, J. Mikko Rautiainen, Aaron Mailman, Chris Gendy, Heikki M. Tuononen

TL;DR
Scientists tried to create a new borylene compound but instead discovered a rare reaction where carbon dioxide inserts into a boron-silicon bond.
Contribution
A rare example of CO2 insertion into a B–Si bond with boron acting as a nucleophile, mimicking transition metal behavior.
Findings
Compound 6 is a rare example of CO2 insertion into a B–Si bond.
Boron functions as a nucleophile in the reaction, mimicking transition metal-mediated carboxylation.
The mechanism of CO2 insertion was analyzed computationally.
Abstract
One-electron reduction of the carbene-stabilized borane (Me2-cAAC)B(Cl)2Si(SiMe3)3, 1, with potassium naphthalenide gave the radical (Me2-cAAC)B(Cl)Si(SiMe3)3, 2. A subsequent one-electron reduction of 2 yielded the dicoordinate borylene (Me2-cAAC)BSi(SiMe3)3, 3, which rapidly underwent intramolecular C–H activation to give cyclo-(Me2-cAAC)B(H)Si(SiMe3)3, 4, irrespective of the employed reaction conditions. Compound 3 could be stabilized as the carbonyl complex (Me2-cAAC)B(CO)Si(SiMe3)3, 5, that gave 4 upon irradiation with a UV light under a CO2 atmosphere. In contrast, the two-electron reduction of 1 under an atmosphere of CO2 yielded a mixture of products of which (Me2-cAAC)B(Cl)(H)C(O)OSi(SiMe3)3, 6, could be separated and structurally characterized. Compound 6 is a rare example of CO2 insertion into a B–E (E = heavier main group element) bond in which boron…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 1
1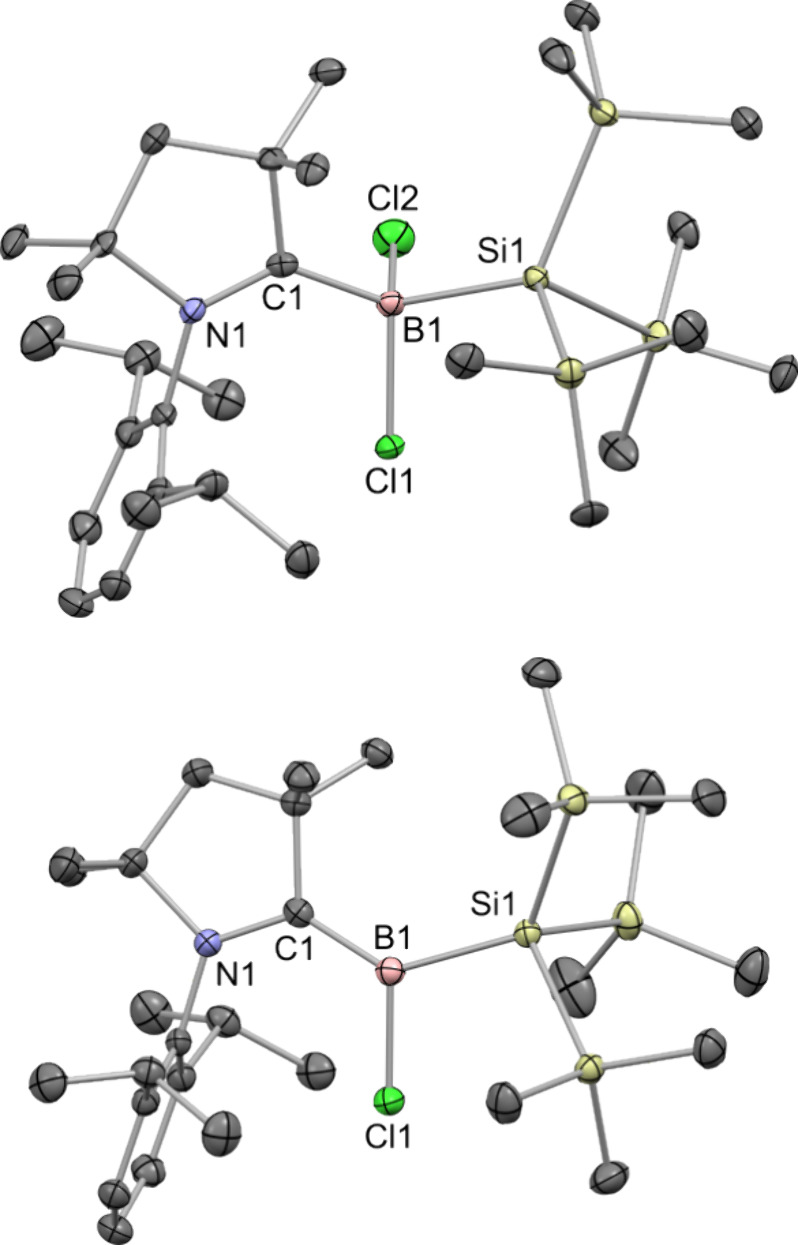 1
1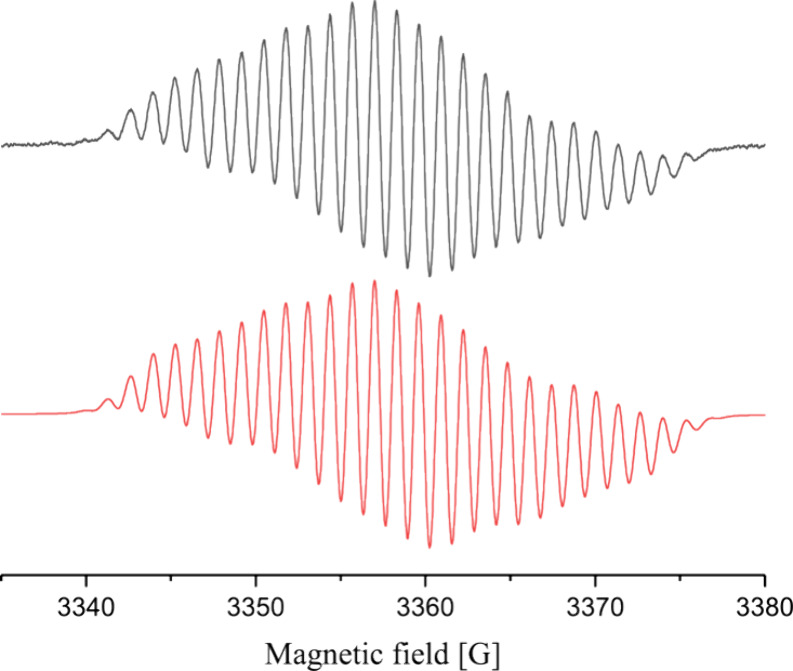 2
2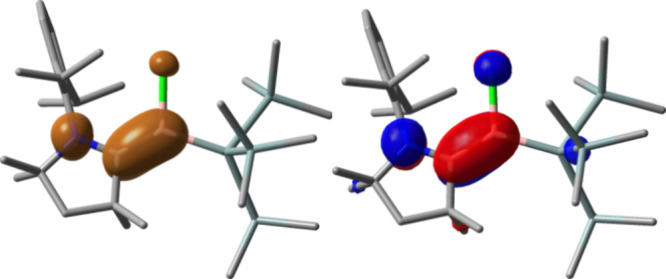 3
3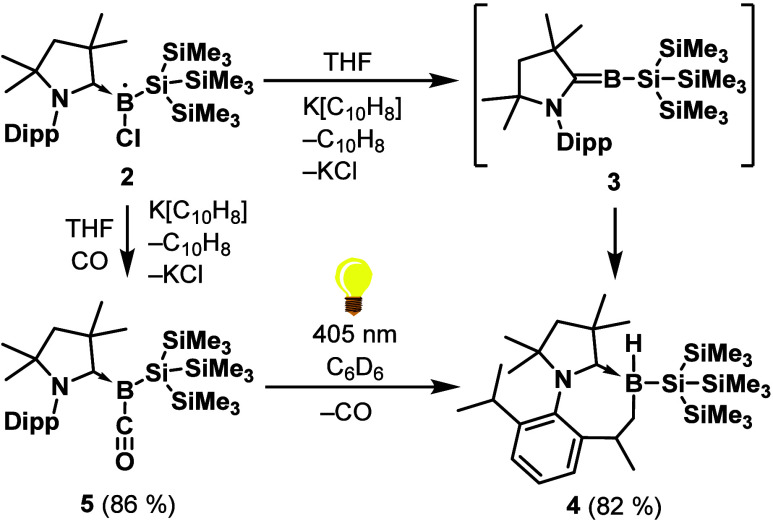 2
2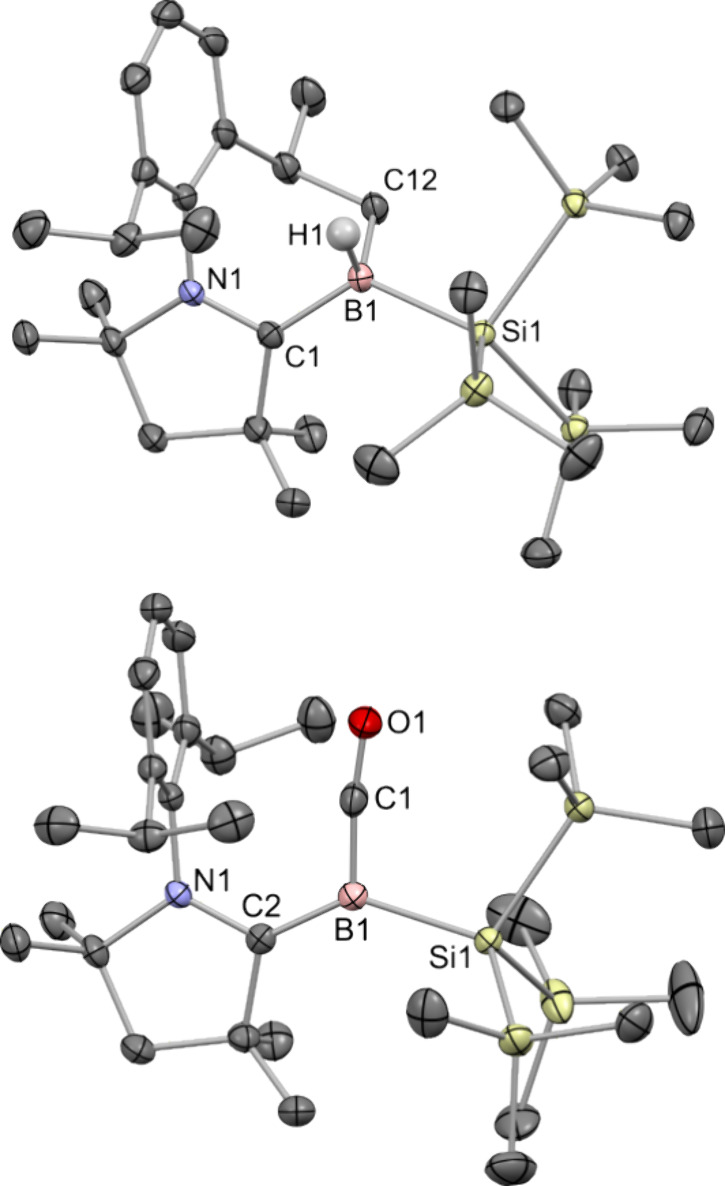 4
4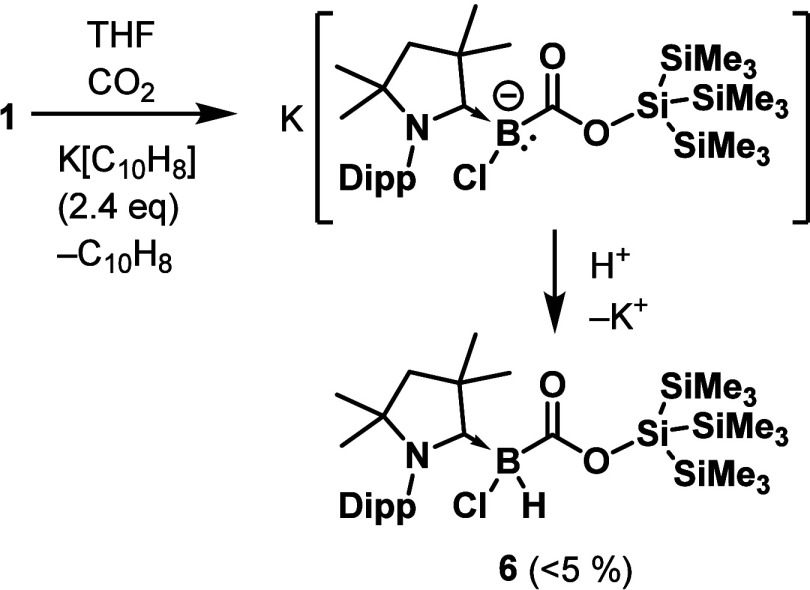 3
3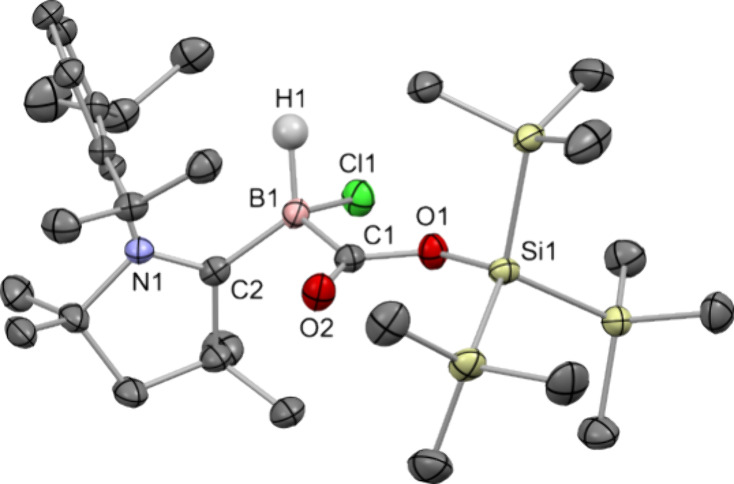 5
5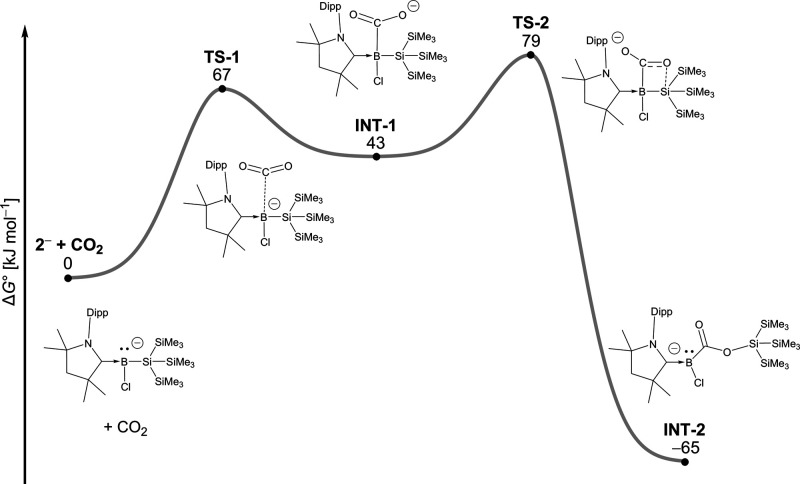 6
6- —European Research Council10.13039/501100000781
- —Jyv?skyl?n Yliopisto10.13039/501100005222
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSynthesis and characterization of novel inorganic/organometallic compounds · Organoboron and organosilicon chemistry · Boron and Carbon Nanomaterials Research
Introduction
Borylenes, general formula **:**B–R, are compounds with an electron-deficient boron center having an organic substituent R, a lone pair of electrons, and two vacant orbitals.? As such, free borylenes are highly reactive species that exist as reaction intermediates and can only be characterized in the gas phase or in low-temperature matrices.? Although complexes of borylenes with transition metals were reported already in the 1990s, ?,? the first example of a borylene stabilized by metal-free reagents, namely, two cyclic(alkyl)amino carbenes (cAACs), was communicated in 2011 by Bertrand and co-workers.? Since that time, a growing number of borylene adducts with one or two auxiliary ligands and a range of organic substituents attached to the boron center have been described.?
The interest in borylenes arises largely from their electronic structure that enables ambiphilic behavior. ?−? ? In this respect, dicoordinate borylenes L–B̈–R (L = ligand) are of significant importance, owing to the balance between stability and reactivity that can be achieved. Specifically, borylenes stabilized by a single Lewis basic ligand can be isolated at room temperature, while their isoelectronic relationship with singlet carbenes allows reactivity akin to transition metals.? For example, stable dicoordinate borylenes are known to activate inert small molecules, such as H_2_,? and readily coordinate an additional ligand, such as CO. ?,?,? Furthermore, as shown by Braunschweig and co-workers, transient dicoordinate borylenes can bind to N_2_ in transition metal-like fashion, allowing its subsequent reduction with potassium graphite.? Fine-tuning the steric bulk of the organic substituent at the borylene unit has even allowed the reductive coupling of two molecules of N_2_.?
Currently, there are only three isolated examples of ligand-stabilized dicoordinate borylenes. ?,?,? Common to all reported systems is the fact that they contain an amino substituent at the borylene unit and employ σ-donating and π-accepting auxiliary carbene ligands for stabilization. Thus, the reactive boron center in these systems is stabilized through a push–pull mechanism, with the shallow bending potential of the allenic C–B–N unit (resonance form CB^–^N^+^) allowing localization of lone pairs and borylene-type reactivity (resonance form C:→B̈–N̈).? In transient dicoordinate borylenes, the substituent at the borylene unit can be of any type, although recent examples have employed sterically bulky carbon-based groups such as 2,3,5,6-tetramethylphenyl (Dur) ?,? or 2,4,6-triisopropylphenyl (Tip).?
Recently, we synthesized germanylidene anions of the type [(Me_2_-cAAC)GeE(SiMe_3_)3]^−^ (Me_2_-cAAC = 1-(2,6-diisopropylphenyl)-3,3,5,5-tetramethylpyrrolidine-2-ylidene; E = Si, Ge) and examined their chemistry with small molecules.? The electronic stabilization afforded by the Me_2_-cAAC ligand in combination with the effective steric protection provided by the hypermetallyl substituent resulted in highly electron-rich nucleophiles that reacted even with weak electrophiles, such as CO_2_. In the current contribution, we report the results of our attempts to use the same ligand-substituent combination to stabilize a dicoordinate borylene (Me_2_-cAAC)BSi(SiMe_3_)3 that would lack an amino substituent on boron. Of key interest was exploring the reactivity of the target species with CO_2_, as only a very few ambiphilic borylenes capable of activating CO_2_ have been reported. Specifically, So and co-workers have communicated CO_2_ capture and functionalization by a bis(N-heterocyclic carbene)-borylene complex,? whereas Wang and collaborators have recently reported the activation and functionalization of CO_2_ with a bis(silylene)-stabilized borylene.? The latter results are of primary interest to our work as the lability of the hypersilyl substituent in (Me_2_-cAAC)BSi(SiMe_3_)3 should allow both single-site as well as cooperative behavior.?
Results and Discussion
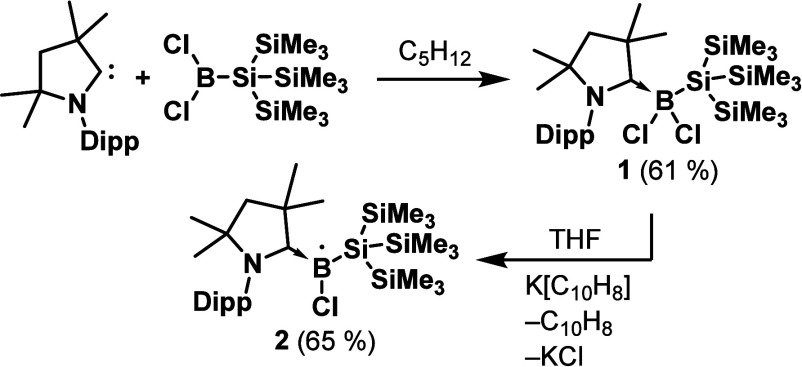
The reaction between freshly prepared Me_2_-cAAC and dichloro{tris(trimethylsilyl)silyl}borane, BCl_2_Si(SiMe_3_)3, in pentane at −78 °C resulted in the precipitation of a colorless product (Me_2_-cAAC)B(Cl)2_Si(SiMe_3)3, 1, which could be isolated as faint-yellow crystals by extraction using hot toluene, followed by crystallization at −20 °C (Scheme). The product was analytically pure and could be stored for months under an inert atmosphere at –30 °C. Analysis of 1 by ^13^C and ^11^B NMR spectroscopies revealed chemical shifts of 217.7 and 1.0 ppm for the carbenic carbon and boron nuclei, respectively, which are comparable to those in related compounds, such as (Me_2_-cAAC)B(Cl)2(Tri) (216.6 and 3.7 ppm for ^13^C and ^11^B, respectively).?
Synthesis of 1 and 2 (Dipp = 2,6-Diisopropylphenyl)
A single-crystal X-ray diffraction analysis of 1 confirmed its structure, showing a four-coordinate boron atom with expected tetrahedral geometry (Figure, top). The B–C bond length in 1 is short, only 1.66(2) Å, and comparable to the lowest end of the range of B–C bond lengths (1.638(3)–1.709(8) Å) reported for 13 related dichlorides of the type (Me_2_-cAAC)B(Cl)2_R in the Cambridge Structural Database (CSD).? In contrast, the B–Si bond length in 1 is rather long, 2.10(1) Å, and comparable to the longest of B–Si(SiMe_3)3 bonds reported to date in the CSD, 2.097(5) Å.?
Single-crystal X-ray structures of 1 (top) and 2 (bottom) with thermal ellipsoids drawn at the 50% probability level. Hydrogen atoms are omitted for clarity.
Reduction of 1 with one equivalent of potassium naphthalenide (K[C_10_H_8_]) resulted in the formation of the radical (Me_2_-cAAC)B(Cl)Si(SiMe_3_)3, 2, which could be recrystallized from cold pentane at −30 °C as big red crystals (Scheme). The product is analytically pure and could be stored for months under an inert atmosphere at −30 °C. A structural analysis by single-crystal X-ray diffraction confirmed the formation of a trigonal planar boron center upon reduction (Figure, bottom). Owing to the decrease in the coordination number at boron, both the B–C and B–Si bonds in 2, 1.522(3) and 2.059(3) Å, respectively, are notably shortened compared to equivalent bonds in 1. The B–C bond length in 2 is statistically equivalent to those in the five known examples of radicals of the type (Me_2_-cAAC)B(Cl)R that span a narrow range from 1.504(3) to 1.524(2) Å. ?,?,?,?
The EPR spectrum of radical 2 (Figure) revealed a symmetric 27-line pattern with g = 2.0016, suggesting the presence of an even number of nuclei with half-integer spins. An excellent (RMSD = 0.0083) simulation of the spectrum could be obtained by using isotropic hyperfine coupling constants to ^11^B (I = 3/2, 3.91 G), ^14^N (I = 1, 6.52 G), ^35^Cl (I = 3/2, 1.23 G), and 6 × ^1^H nuclei (I = 1/2, 1.35–1.40 G), and isotopologs containing ^10^B and/or ^37^Cl nuclei with appropriate couplings. DFT-predicted hyperfine coupling constants agree well with the data inferred from the experimental spectrum: A(^11^B) = 4.11 G, A(^14^N) = 4.62 G, A(^35^Cl) = 1.64 G, and 6 × A(^1^H) = 0.91–1.60 G. A population analysis of the calculated spin density showed that 2 is a cAAC-stabilized boron radical with α-spin density mostly divided between B (39%), C (33%), N (24%), and Cl (4%) (Figure). For comparison, the related radical (Me_2_-cAAC)B(Cl)(Dur) has the α-spin density largely on the carbenic carbon (50%), with the reminder split almost equally between B (27%) and N (24%).? These results suggest that 2 could display boron-centered reactivity. Nevertheless, the radical proved to be a very stable species, and attempts to react it with a range of different substrates were unsuccessful. We note that as a crystalline solid, 2 reacts very slowly even with air at ambient temperature.
EPR spectrum of 2 measured at 273 K in n-hexane (top, black) and simulated using Voigtian line shape with 0.59 and 0.39 G fwhm for Gaussian and Lorenzian components, respectively (bottom, red). For full details of the simulation, see the main text and the Supporting Information (SI).
DFT-calculated spin density map (density value ± 0.004; left) and singly occupied molecular orbital (isovalue ± 0.05; right) of 2. Hydrogen atoms are omitted for clarity.
Reduction of 2 with one equivalent of K[C_10_H_8_] failed to yield the target dicoordinate borylene (Me_2_-cAAC)BSi(SiMe_3_)3, 3. Instead, conversion to the intramolecularly cyclized product cyclo-(Me_2_-cAAC)B(H)Si(SiMe_3_)3, 4, was observed irrespective of the employed reaction conditions (Scheme). The identity of 4 was confirmed via crystallographic analysis, which revealed that a C–H activation of an isopropyl group on the cAAC ligand has taken place, giving a tetrahedral boron center with a B–H bond length of 1.16(1) Å (Figure, top). In agreement with the solid-state structural data, the FTIR spectrum of 4 displayed a characteristic B–H stretch at 2310 cm^–1^, while its ^11^B NMR spectrum showed splitting of the peak at −25.9 ppm into a doublet (^1^ J BH = 89.9 Hz). Similar C–H activation has been reported for other dicoordinate borylenes employing the Me_2_-cAAC ligand, giving either an analogous B–H product? or a related C–H species that forms via subsequent hydride migration. ?,? Related C–H activation pathways involving the Et_2_-cAAC and Cy-cAAC ligands are also known. ?−? ? ?
Synthesis of 4 and 5, and the Photolytic Conversion of 5 to 4 (Dipp = 2,6-Diisopropylphenyl)
Single-crystal X-ray structures of 4 (top) and 5 (bottom) with thermal ellipsoids drawn at the 50% probability level. C–H hydrogen atoms are omitted for clarity.
A DFT analysis of 4 showed that the observed B–H product is favored over its C–H tautomer by 15 kJ mol^–1^. Furthermore, the Gibbs energy of activation for the conversion of 3 to 4 was calculated to be 69 kJ mol^–1^. This is on par with the barriers reported by Braunschweig and co-workers for transient dicoordinate borylenes of the type (Me_2_-cAAC)BR (R = Dur, Tip, Mes = 1,3,5-trimethylphenyl).? In contrast, the activation energies calculated for intramolecular cyclization of (Me_2_-cAAC)BN(SiMe_3_)2 and (Me_2_-cAAC)B(TMP) (TMP = 2,2,6,6-tetramethylpiperidyl) are 124 and 156 kJ mol^–1^, respectively, in good agreement with the electronic stabilization provided by the amino substituents and the stability of both borylenes as free species. Consequently, the steric protection offered by the Si(SiMe_3_)3 substituent is unable to compensate for the inherent electronic instability of the dicoordinate boron center in 3, resulting in the observed intramolecular cyclization and formation of 4.
Because 3 could not be isolated, its trapping as a carbonyl complex (Me_2_-cAAC)B(CO)Si(SiMe_3_)3, 5, was attempted by performing the reduction of 2 under an atmosphere of carbon monoxide (Scheme) analogously to what has been reported for related transient dicoordinate borylenes. ?,?,?,? Gratifyingly, 5 could be isolated in good yield as a bright-yellow crystalline product whose composition was confirmed by single-crystal X-ray analysis and FTIR spectroscopy (Figure, bottom). These revealed a B–CO moiety with B–C and C–O distances of 1.456(3) and 1.164(3) Å, respectively, along with a characteristic CO stretch at 1950 cm^–1^ (cf. 1.469(2) and 1.158(2) Å, and 1942 cm^–1^ reported for (Me_2_-cAAC)B(Dur)).?
With carbonyl complex 5 at hand, it could be used as an in situ source of 3 by cleaving the B–CO bond with UV light. When the photolytic reaction was performed under an argon atmosphere at room temperature, a quantitative conversion of 5 to 4 took place within 17 h, as evidenced by NMR spectroscopy (for details, see the SI). Hence, photolysis of 5 was performed under an atmosphere of carbon dioxide. Unfortunately, all attempts to realize reactivity between CO_2_ and the in situ formed 3 were thwarted by the facile intramolecular cyclization of the latter, giving 4 as the only product. Consequently, the reactivity between 3 and CO_2_ was approached from another perspective by performing the two-electron reduction of 1 with an excess of K[C_10_H_8_] under an atmosphere of CO_2_ at −78 °C (Scheme). It was hoped that under such reductive conditions, fleeting intermediates formed by CO_2_ and in situ generated 3 could be stabilized by electron transfer before intramolecular cyclization to 4 had sufficient time to proceed.
Synthesis of 6 by the Reduction of 1 under an Atmosphere of CO2 (Dipp = 2,6-Diisopropylphenyl)
NMR spectroscopy showed that the two-electron reduction of 1 under an atmosphere of CO_2_ gave a mixture of compounds, including 4, from which a small amount of crystalline 6 could be separated. To our surprise, a subsequent single-crystal X-ray structural analysis (Figure) showed that 6 is not a reductively stabilized adduct between CO_2_ and 3. Instead, it appears to be a product from the insertion of a molecule of carbon dioxide into the B–Si bond of a boryl anion intermediate, [(Me_2_-cAAC)B(Cl)Si(SiMe_3_)3]^−^, 2 ^ – ^. Related boryl anions of the type [(Me_2_-cAAC)BCl_2_]^−^ and [(Me_2_-cAAC)B(Cl)H]^−^ have been observed as reactive, low-yield intermediates in the synthesis of chloroborylenes (Me_2_-cAAC)B(Cl)R and dihydrodiborenes [(Me_2_-cAAC)BH]2, respectively, by reduction of appropriate precursors.? It is, therefore, likely that a similar anionic intermediate is formed in the reduction of 1, which reacts with CO_2_ through insertion. The structurally characterized product 6, (Me_2_-cAAC)B(Cl)(H)C(O)OSi(SiMe_3_)3, forms upon subsequent proton capture from an unknown source (presumably the Si(SiMe_3_) group of an adjacent anion). The hydrogen atom in 6 could be located from the difference Fourier map, though the B–H distance determined through this route, 1.32(3) Å, is associated with a high uncertainty and is markedly longer than the calculated B–H bond length (1.205 Å).
Single-crystal X-ray structure of 6 with thermal ellipsoids drawn at the 50% probability level. C–H hydrogen atoms are omitted for clarity.
In CO_2_ activation mediated by frustrated Lewis pairs, the electrophile, such as boron in boranes, captures CO_2_ via O-coordination, whereas the η^1^-CO_2_-κC binding mode is observed for the nucleophile, such as phosphorus in phosphines.? In contrast, the insertion of CO_2_ into the B–Si bond in 6 has taken place in an “inverse” manner with boron acting as the nucleophile and silicon as the electrophile. This is reminiscent of transition metal-mediated carboxylation reactions in which CO_2_ inserts into a metal–nucleophile bond.? In the realm of related main group compounds, similar reactivity has been observed for a C–Si bond in a (phosphino(trimethylsilyl)methyl)pyridine ligand? and for a N–Si bond in an N-heterocyclic iminosilane.?
Given the rarity of examples of CO_2_ activation mediated by B–E compounds (E = heavier main group element) in which boron is nucleophilic,? the mechanism for the formation of 6 was examined in detail using DFT. In agreement with experimental observations, analyses of reactivity between CO_2_ and 1, 2, or 3 failed to identify low energy pathways through which an insertion product 6 could be formed. In contrast, the reaction between the purported boryl anion 2 ^–^ and CO_2_ was found to be facile, giving a labile η^1^-CO_2_ intermediate INT-1 (Figure). This intermediate can either dissociate or undergo a subsequent intramolecular silyl group migration from boron to oxygen, giving the μ_2_,η?-CO_2_ product INT-2 with an overall Gibbs energy change ΔG° = –65 kJ mol^–1^ in the gas phase; a simple proton transfer generates 6 from INT-2. The pathway in Figure is mechanistically identical to that reported for the insertion of carbon dioxide into an N-heterocyclic iminosilane,? but the relative stabilities of the reaction products differ markedly. Specifically, the activation barrier for the reverse deinsertion is 144 kJ mol^–1^ for INT-2, whereas it is only 73 kJ mol^–1^ for N-heterocyclic iminosilanes.? Thus, the insertion of CO_2_ into a N–Si bond in N-heterocyclic iminosilanes is reversible in solution at room temperature.
Calculated mechanism for the insertion of CO2 into the B–Si bond. Relative Gibbs energies refer to gas phase calculations at room temperature.
The poor yield of 6 can be rationalized based on the instability of the key boryl anion intermediate 2 ^ – ^ along with the fact that the reaction landscape of INT-1 has a slight preference for dissociation (TS-1) over insertion (TS-2). To increase the synthetic yield of 6, the in situ two-electron reduction of 1 was performed by using an excess of [K(18c6)][C_10_H_8_] as encapsulation of the cation should decrease the likelihood of chloride abstraction from 2 ^ – ^, thereby allowing its reactivity with CO_2_ and subsequent generation of INT-2 and, ultimately, 6. Unfortunately, no improvement in the yield of 6 was observed experimentally.
Conclusions
One-electron reduction of the Me_2_-cAAC-stabilized precursor 1 gave the boron-centered radical 2 that proved to be a surprisingly stable species and unreactive toward a range of substrates. A subsequent one-electron reduction of 2 yielded the cyclized product 4 through facile intramolecular C–H activation involving the dicoordinate borylene intermediate 3. While the sterically demanding hypersilyl ligand did not allow the isolation of compound 3, it could be trapped as photocleavable carbonyl complex 5. Subsequent reactions of carbon dioxide with 3, generated either in situ from 5 by UV irradiation or from 1 by reduction with two equivalents of K[C_10_H_8_], did not yield the anticipated activation product, presumably owing to the low barrier associated with the formation of 4. However, the latter reaction gave a mixture of products, from which 6 could be separated. A structural characterization of 6 by X-ray crystallography revealed that an insertion of CO_2_ into a transient boryl anion intermediate had taken place, in analogy with transition metal-mediated carboxylation. Similar reactivity has been reported for compounds with related C–Si and N–Si interactions, with 6 completing the series with its B–Si bond. Given the scarcity of characterized examples of insertion of CO_2_ into B–E functionalities (E = heavier main group element) in which boron acts as the nucleophile, the results obtained herein highlight the interesting possibility of using suitable three-coordinate boryl anions for the activation of carbon dioxide. Such investigations are currently ongoing in our group.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Soleilhavoup M.Bertrand G.Borylenes: An Emerging Class of Compounds Angew. Chem., Int. Ed.20175635102821029210.1002/anie.20170515328577325 · doi ↗ · pubmed ↗
- 2Braunschweig H.Kollann C.Englert U.Synthesis and Structure of the First Terminal Borylene Complexes Angew. Chem., Int. Ed.199837223179318010.1002/(SICI)1521-3773(19981204)37:22<3179::AID-ANIE 3179>3.0.CO;2-Z 29711330 · doi ↗ · pubmed ↗
- 3Braunschweig H.Transition Metal Complexes of Boron Angew. Chem., Int. Ed.19983713–141786180110.1002/(SICI)1521-3773(19980803)37:13/14<1786::AID-ANIE 1786>3.0.CO;2-C · doi ↗
- 4Kinjo R.Donnadieu B.Celik M. A.Frenking G.Bertrand G.Synthesis and Characterization of a Neutral Tricoordinate Organoboron Isoelectronic with Amines Science 2011333604261061310.1126/science.120757321798945 · doi ↗ · pubmed ↗
- 5Dahcheh F.Martin D.Stephan D. W.Bertrand G.Synthesis and Reactivity of a CAAC–Aminoborylene Adduct: A Hetero-Allene or an Organoboron Isoelectronic with Singlet Carbenes Angew. Chem., Int. Ed.20145348131591316310.1002/anie.20140837125267591 · doi ↗ · pubmed ↗
- 6Arrowsmith M.Auerhammer D.Bertermann R.Braunschweig H.Celik M. A.Erdmannsdörfer J.Krummenacher I.Kupfer T.From Borane to Borylene without Reduction: Ambiphilic Behavior of a Monovalent Silylisonitrile Boron Species Angew. Chem., Int. Ed.20175637112631126710.1002/anie.20170556128640395 · doi ↗ · pubmed ↗
- 7Dewhurst R. D.LégaréM.-A.Braunschweig H.Towards the Catalytic Activation of Inert Small Molecules by Main-Group Ambiphiles Commun. Chem.2020311410.1038/s 42004-020-00371-436703344 PMC 9814040 · doi ↗ · pubmed ↗
- 8Witte R.Arrowsmith M.Lamprecht A.Schorr F.Krummenacher I.Braunschweig H.C–C and C–N Bond Activation, Lewis-Base Coordination and One- and Two-Electron Oxidation at a Linear Aminoborylene Chem. – Eur. J.20232916 e 20220366310.1002/chem.20220366336562195 · doi ↗ · pubmed ↗