Single-stranded DNA drives σ subunit loading onto mycobacterial RNA polymerase to unlock initiation-competent conformations
Rishi Kishore Vishwakarma, Nils Marechal, Zakia Morichaud, Mickaël Blaise, Emmanuel Margeat, Konstantin Brodolin

TL;DR
This study reveals how single-stranded DNA helps the σ subunit load onto mycobacterial RNA polymerase, enabling transcription initiation.
Contribution
The study identifies a novel mechanism where ssDNA guides RNA polymerase maturation and σ subunit regulation.
Findings
−10 ssDNA induces σ subunit loading onto RNA polymerase channels.
ssDNA triggers RNAP clamp closure and unswiveling necessary for transcription initiation.
σ subunit acts as a regulator of RNA polymerase conformational dynamics.
Abstract
Initiation of transcription requires the formation of the “open” promoter complex (RPo). For this, the σ subunit of bacterial RNA polymerase (RNAP) binds to the nontemplate strand of the −10 element sequence of promoters and nucleates DNA unwinding. This is accompanied by a cascade of conformational changes on RNAP, the exact mechanics of which remains elusive. Here, using single-molecule Förster resonance energy transfer and cryo-electron microscopy, we explored the conformational landscape of RNAP from the human pathogen Mycobacterium tuberculosis upon binding to a single-stranded DNA (ssDNA) fragment that includes the −10 element sequence (−10 ssDNA). We found that like the transcription activator RNAP-binding protein A, −10 ssDNA induced σ subunit loading onto the DNA/RNA channels of RNAP. This triggered RNAP clamp closure and unswiveling that are required for RPo formation and RNA…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
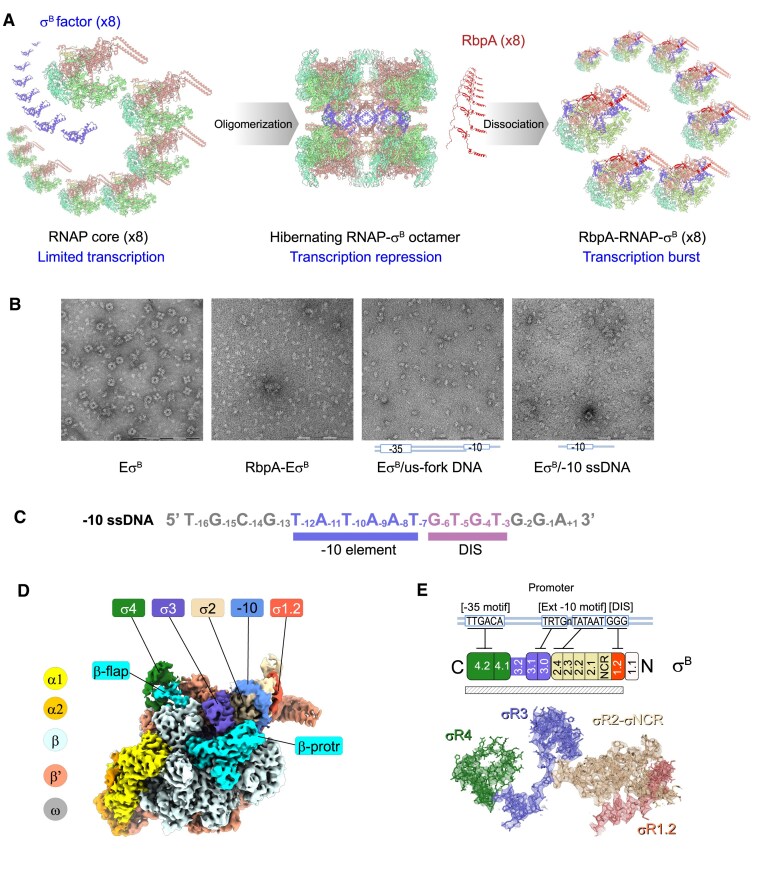 Figure 1
Figure 1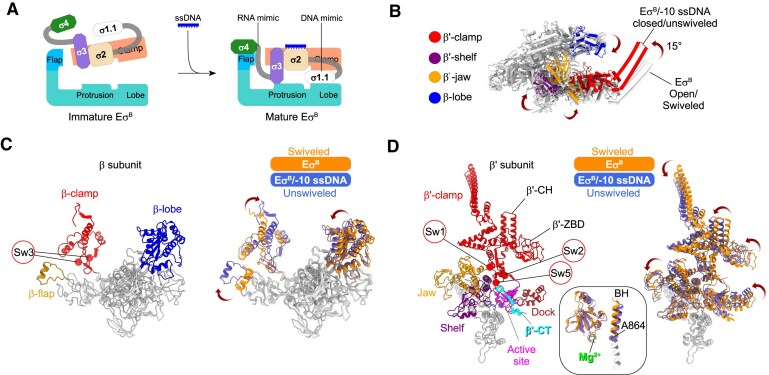 Figure 2
Figure 2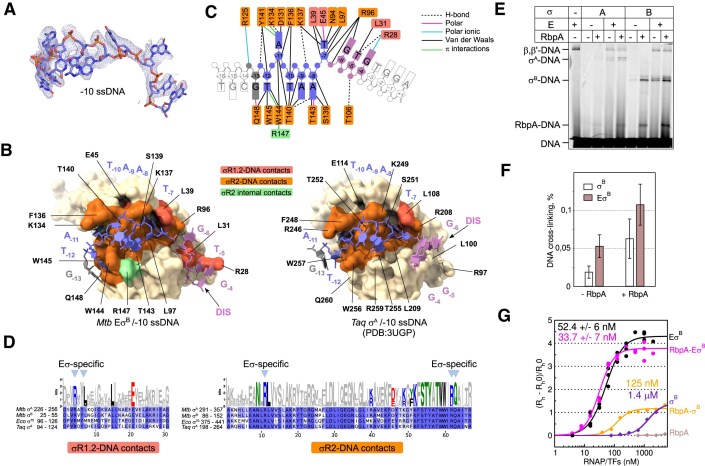 Figure 3
Figure 3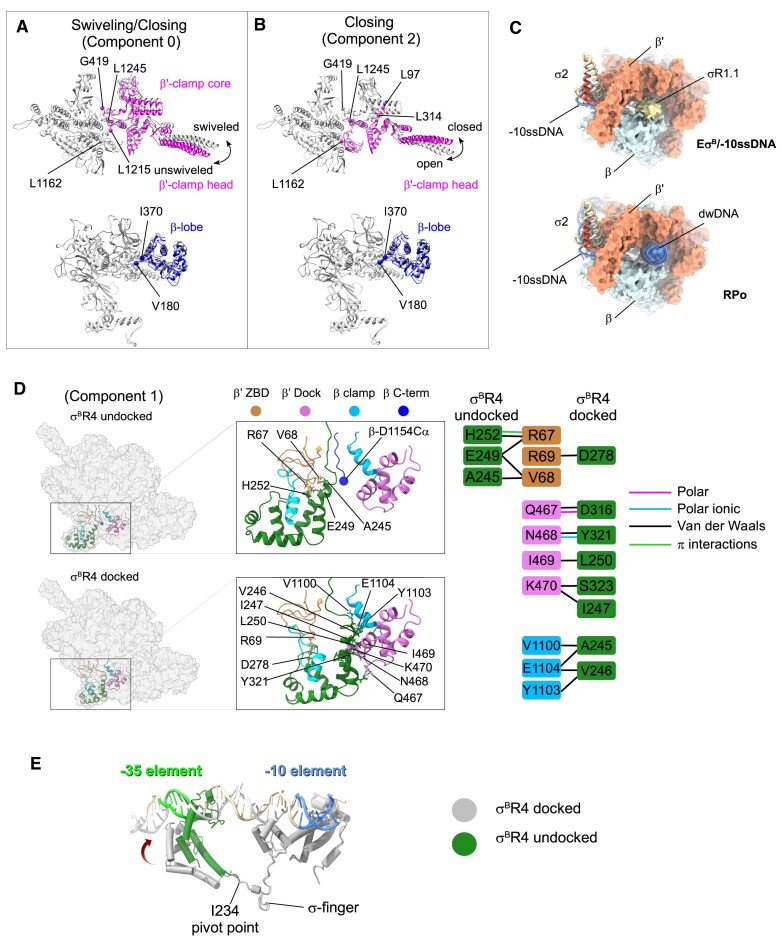 Figure 4
Figure 4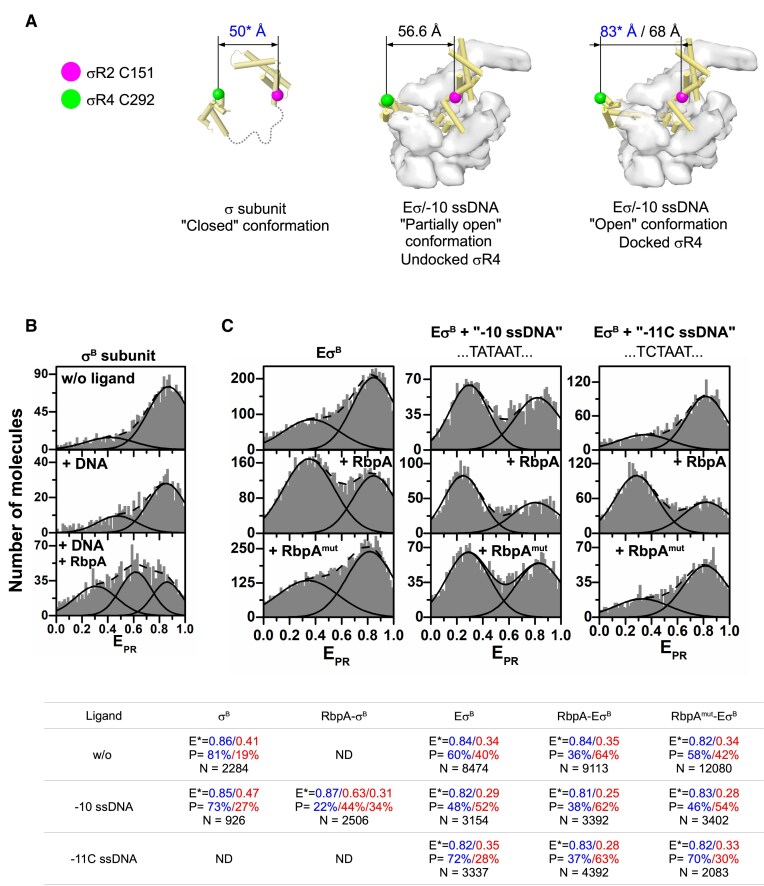 Figure 5
Figure 5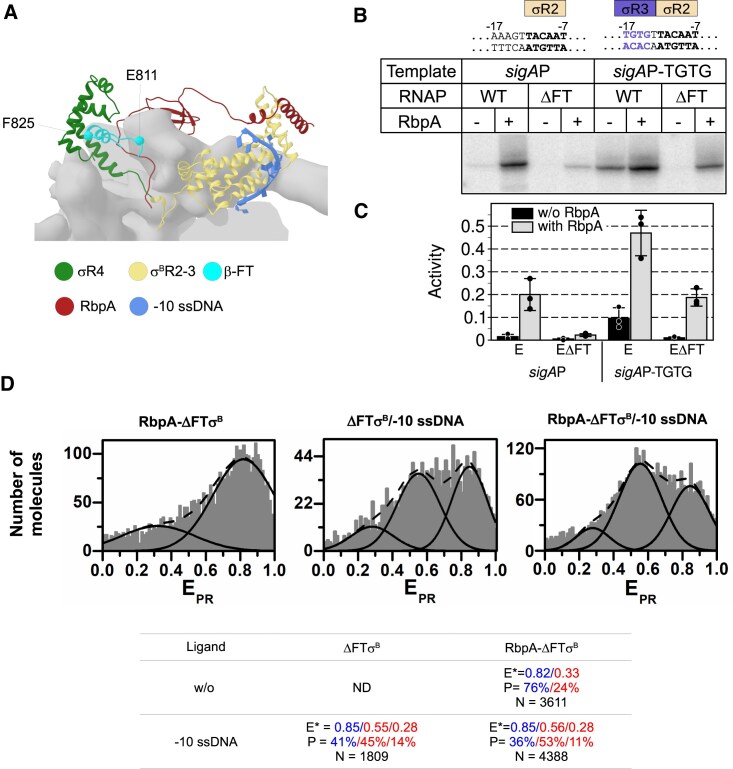 Figure 6
Figure 6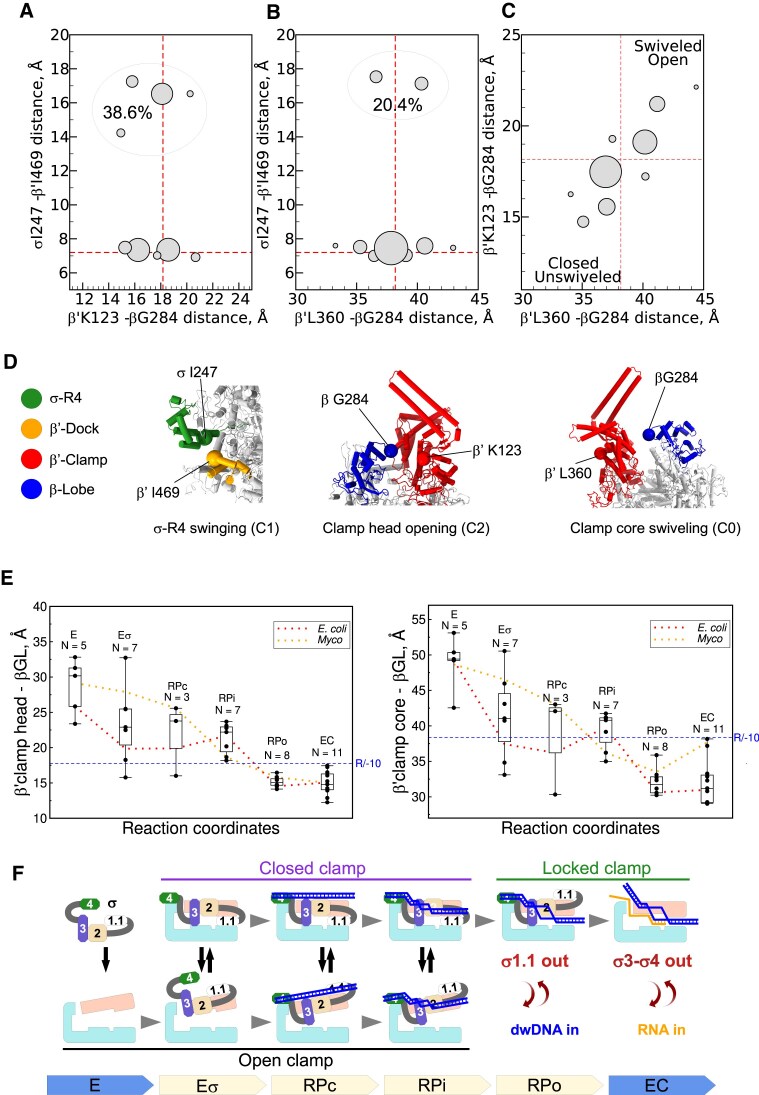 Figure 7
Figure 7| Conformational states | Moving domain | Rotation angle | Distance between mass centers |
|---|---|---|---|
| EσB octamer versus EσB/−10 ssDNA (consensus I) | Clamp core | 15.27° | 10.5 Å |
| β-lobe1 | 3.59° | 2.31 Å | |
| EσB/−10 ssDNA unswiveled versus swiveled (3DVA component 0) | Clamp core | 9.38° | 5.69 Å |
| β-lobe1 | 3.68° | 1.74 Å | |
| EσB/−10 ssDNA open versus closed (3DVA component 2) | Clamp head /β′i1 | 13.57° | 5.03 Å |
| β-lobe1 | 3.71° | 1.95 Å | |
| EσB/−10 ssDNA | σR4 | 31.63° | 10.02 Å |
- —French National Research Agency10.13039/501100001665
- —Instruct-ERIC
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRNA and protein synthesis mechanisms · Bacterial Genetics and Biotechnology · Bacteriophages and microbial interactions
Introduction
Transcription is the first step in gene expression that is required to interpret the information encoded in genomic DNA. To initiate transcription, all cellular RNA polymerases (RNAPs) must recognize promoter DNA motifs, locally melt duplex DNA, and unwind the transcription start site. Duplex DNA is an energetic barrier for transcription initiation and requires the application of mechanical force by RNAPs. In bacteria, transcription initiation is performed by the RNAP holoenzyme (Eσ) assembled from the RNAP core (E; subunits α2ωββ′) and the σ subunit to mediate promoter recognition and DNA melting [1, 2]. The σ subunits are classified into four groups according to their role and number of structural domains (numbered 1.1 to 4) [3]. Group I includes the housekeeping σ subunit [σ^70^ in Escherichia coli (Eco) and σ^A^ in Mycobacterium tuberculosis (Mtb) and Thermus aquaticus (Taq)] and controls transcription of most genes during exponential growth. Group II σ subunits (Eco σ^38^ and Mtb σ^B^) have a shorter region 1.1 compared with group I and are implicated in the stress response and stationary phase growth [3, 4]. During transcription initiation, σ region 2 (σR2) and region 4 (σR4) recognize the promoter −10 element sequence (T_−12_A_−11_T_−10_A_−9_A_−8_T_−7_) and −35 element sequence (TTGACA), respectively, to form the closed promoter complex (RPc). RPc spontaneously isomerizes to an open complex (RPo) through several structurally distinct intermediates (RPis) [5–7]. In RPo, RNAP is ready to initiate RNA synthesis and forms a stable elongation complex after the synthesis of 11–14-nt RNA. The σ subunit regions σR1.1, σR3, and σR4 occupy the DNA- and RNA-binding channels of the RNAP core and should be displaced in a step-wise fashion by the DNA template (σR1.1 and σR3) and by nascent RNA (σR3 and σR4) during transcription initiation [8].
Binding of σR2 to single-stranded DNA (ssDNA) bearing the −10 element sequence (−10 ssDNA) is a key event that triggers promoter melting. σR2 captures the −11A base flipped out of the duplex DNA [9, 10]. This results in local DNA melting and then unwinding of ∼13 bp of DNA duplex [11, 12]. Base-flipping is the universal mechanism for nucleation of promoter melting by all classes of σ factors [13–15]. Biochemical studies showed that the free σ subunit and σR2 alone can recognize the −10 motif and bind to −10 ssDNA with low affinity [9, 16]. Binding of σR2 to the β′ subunit clamp coiled-coil, also called clamp helices (β′-CH), triggers a cryptic conformational switch in σR2 that increases its affinity for −10 ssDNA [16–19]. Although σR2 can bind to −10 ssDNA, neither free σ nor σR2 alone can melt promoter DNA duplex without the RNAP core [20]. It has been suggested that the opening–closing dynamics of the RNAP clamp and β subunit lobe (called RNAP pincers) play an essential role in RPo formation by RNAPs in all the tree domains of life [21–26]. Single-molecule Förster resonance energy transfer (smFRET) studies of Eco RNAP showed that the clamp mainly adopts an open state in the RNAP core and Eσ^70^ holoenzyme, and a closed state in RPo and after promoter escape during elongation [27–30]. In Eσ, the gap between σR2 bound to clamp and β-lobe (10–12 Å) is narrower than the double-stranded DNA (dsDNA) helix diameter (20 Å). Therefore, as σR2 obstructs dsDNA access to the RNAP active site cleft [23, 31], the clamp should be opened to allow dsDNA entry and then closed over DNA to hold it in the cleft. Conversely, entry of ssDNA (diameter <10 Å) does not necessitate clamp opening [32]. The importance of the clamp opening–closing dynamics for RPo formation is supported by studies on antibiotics that bind to the RNAP switch regions [33, 34]. Specifically, fidaxomicin inhibits transcription initiation by blocking the clamp in the open state [11, 34–36]. Conversely, myxopyronin and corallopyronin block the clamp in the closed state [32, 33, 37]. Although clamp dynamics seem to be important for RPo formation, no clear causal relationship has been established between clamp dynamics and promoter melting [38]. Structural studies of promoter melting intermediates formed by Eco Eσ^70^ [11, 12, 39] and Mtb Eσ^A^ [32, 40] suggest that nucleation of the duplex −10 element DNA melting takes place outside the RNAP cleft and may require clamp closure [11]. Conversely, entry of the downstream DNA duplex (dwDNA) into the DNA channel requires clamp opening. Unwinding of the transcription start site occurs in the RNAP cleft after dwDNA entry and requires a transient increase in the distance between the β′ switch 2 and β fork-loop 2 that restricts ssDNA access to the active site [32]. In the alternative view, unwinding occurs outside the active site cleft, followed by the ssDNA loading inside [31, 41]. Indeed, smFRET studies showed that the clamp remains in the closed conformation during promoter binding and unwinding, suggesting no transient clamp opening during RPo formation [28]. Moreover, neither model of RPo formation can explain biochemical data showing that Eco σR2–R3 and β′ subunit clamp domain (aa 1–314) are sufficient to melt supercoiled DNA duplex [20]. As in this “minimal” system clamp closure is irrelevant, it is not clear how DNA unwinding occurs.
In Mtb, RPo formation by RNAPs that contain the principal σ^A^ or principal-like σ^B^ subunits is regulated by RNAP-binding protein A (RbpA), essential for bacterial growth [42–45]. Our studies depicted RbpA as a transcription factor that stabilizes σ^A^ and σ^B^ interactions with the RNAP core [43, 44], stabilizes RPo, and decreases the energetic barrier for promoter DNA melting [43, 46]. RbpA interacts with the β′ subunit Zn^2+^-binding domain (β′-ZBD) and with the nonconserved region (NCR) of σ (σ-NCR), which is regulated by lineage-specific transcription factors [47–49]. Biochemical, biophysical, and structural studies suggest that RbpA acts as a σ-loader, inducing a σ conformational change that results in σR2 and σR4 stretching over the RNAP core surface to match the distance between the −10 and −35 elements [49, 50]. We use the term “RNAP maturation” to describe the process of σR4 loading into RNA channel in order to highlight the fact that RNAP undergoes transition from a promoter binding-deficient conformation to a fully active conformation. In the absence of RbpA, the Eσ^B^ holoenzyme (but not Eσ^A^) oligomerizes to an octamer [51] in which the RNAP clamp is captured in a fully opened conformation, similar to the one stabilized by fidaxomicin. We previously proposed [51] that the Mtb Eσ^B^ octamer may represent a hibernating form of RNAP, analogous to the dimeric forms of eukaryotic PolI and PolII [52–54]. RbpA binding, which induces σ^B^ remodeling, also leads to clamp closure [35] and octamer dissociation [51] (Fig. 1A). Here, we used Mtb Eσ^B^ as a model system to explore the link between clamp dynamics, −10 element recognition, and σ remodeling. We found that the −10 ssDNA fragment is sufficient to induce huge RNAP structure rearrangements, leading to the Eσ^B^ holoenzyme release from a conformational lock. Our results suggest that ssDNA, together with the σ subunit, acts as driver of RPo formation by triggering clamp closure.
Structure of the M. tuberculosis EσB holoenzyme in complex with −10 ssDNA. (A) Cartoon depicting the Mtb EσB holoenzyme assembly followed by oligomerization into an octamer and its regulation by RbpA. RbpA-induced EσB octamer dissociation results in the increase of the active RbpA–EσB concentration, leading to a transcription burst [51]. (B) Mtb EσB octamer disassembly induced by RbpA and by −10 ssDNA. Negatively stained electron microscopy (EM) images of Mtb RNAP holoenzyme octamers (EσB), EσB monomers in complex with RbpA (RbpA–EσB), Mtb EσB monomers in complex with us-fork DNA spanning the promoter positions −42/−3 (EσB/us-fork DNA), and EσB in complex with the −10 ssDNA (EσB/−10 ssDNA). Scale bar = 200 nm. (C) Sequence of the 17-mer DNA oligonucleotide derived from the lacUV5 promoter (−10 ssDNA). The −10 sequence element and discriminator (DIS) element, which determines whether promoter is subject to stringent control, are underlined. (D) Overall cryo-electron microscopy (cryo-EM) map of the Mtb EσB/−10 ssDNA complex (consensus I map). RNAP subunits are color-coded as indicated on the left. β-protrusion (β-protr) and β-flap are in cyan. Regions/domains of the σB subunit and −10 ssDNA (-10) are color-coded as indicated on the top. (E) Cryo-EM density (shown as mesh) and molecular model of the resolved segment of the σB subunit. The cartoon shows the σB organization (numbered regions and subregions) and its interaction with promoter elements. N-terminus (N) on the right and C-terminus (C) on the left. The subregions inside σ domains are numbered. The hatched rectangle shows the resolved σB segment (residues 24–323). The σB subunit regions/domains σR1.2 (aa 24–55), σ-NCR (aa 56–86), σR2 (aa 87–163), σR3 (aa 164–240), and σR4 (aa 241–323) are color-coded as indicated.
Materials and methods
Proteins and DNA templates
The M. tuberculosis RNAP core (harboring the C-terminal 6×His-tag on rpoC) and its mutant (ΔFT), in which amino acids 811–825 of the β subunit were deleted, were expressed in BL21 DE3 E. coli cells transformed with the pMR4 plasmid and purified as described before [43, 51]. The 6×His-tagged σ^A^, σ^B^, σ^B^-Cys151/292 mutant, and RbpA were expressed in BL21 DE3 E. coli cells and purified as described before [43, 50]. Mutant RbpA-R88,89A was constructed using the Quick Change Lightening site-directed mutagenesis kit (Agilent). DNA oligonucleotides, purified by high-performance liquid chromatography (Supplementary Table S1), were purchased from Sigma–Aldrich.
Protein labeling and smFRET measurements
Random labeling of the σ^B^ subunit with the DY547P1 and DY647P1 fluorescent dyes at Cys151/Cys292 was performed as described before [50]. The fluorescent dye derivatives DY547P1-maleimide (donor, D) and DY647P1-maleimide (acceptor, A) were purchased from Dyomics GmbH. The double-labeled σ^B^ subunit at 25 pM was prepared in filtered (0.1 μm) FRET buffer [20 mM Tris–HCl (pH 7.9), 150 mM NaCl, 5 mM MgCl_2_, 5% glycerol, bovine serum albumin (BSA; 0.1 mg/ml)]. smFRET measurements were performed using a home-built confocal PIE-MFD (Multiparameter Fluorescence Detection with Pulsed Interleaved Excitation) microscope, as described before [50, 55]. Briefly, samples were placed in clear nonbinding surface 384-well plates (Corning) and acquisitions were performed for 1.5–3 h at 22°C. Data were analyzed with the “Software Package for Multiparameter Fluorescence Spectroscopy, Full Correlation and Multiparameter Fluorescence Imaging” (https://www.mpc.hhu.de/software/mfd-fcs-and-mfis) [56]. A single-molecule event was defined as a burst containing at least 30 photons. Then the relevant species containing donor and acceptor (D-A) were extracted by selecting only the molecules that satisfy the following criteria: (i) number of photons detected in the acceptor channel upon acceptor excitation > 20; (ii) excited state lifetime of the acceptor upon acceptor excitation 0.55 ns < τA < 2.65 ns; and (iii) stoichiometry ratio S > 0.4. Photobleached molecules were eliminated as described [57]. For each photon burst, the apparent D-A FRET efficiencies (EPR) were calculated with the equation EPR = (IA)/(ID+ IA), where IA and ID are the intensities detected on the donor and acceptor channels, respectively, and plotted. The EPR histograms were build using the OriginPro software.
Run-off transcription assays
Briefly, 100 nM of RNAP core and 300 nM of the σ subunit were incubated in transcription buffer (TB) [20 mM Tris–HCl (pH 7.9), 50 mM NaCl, 5 mM MgCl_2_, 0.5 mM dithiothreitol (DTT), 0.1 mM ethylenediaminetetraacetic acid, 1 μM ZnCl_2_, 5% glycerol] at 37°C for 10 min. When indicated, RbpA was added to 300 nM. The reaction mixtures were incubated with the sigAP and sigAP-TGTG derivative (40 nM) promoters at 37°C for 10 min. Transcription was initiated by adding ATP, GTP, and CTP to a final concentration of 25 μM/each, 3 μCi of [a-^32^P] UTP (PerkinElmer Life Sciences) or [a-^33^P] UTP (Hartmann Analytic), and 1 μM UTP and was carried out at 37°C for 5 min.
DNA–protein cross-linking by formaldehyde
Cross-linking reactions and analysis of cross-linked complexes were performed as described before [58] with the following modifications. Briefly, the indicated combinations of RNAP core at 400 nM, σ^A^, σ^B^ at 600 nM, and RbpA at 400 nM were mixed in TB and incubated at 37°C for 10 min. The Cy5-labeled DNA oligonucleotide was added to 50 nM and incubated at 22°C for 20 min. Formaldehyde was added to 0.1%, and cross-linking was performed for 30 s. Reactions were stopped by adding sodium dodecyl sulfate (0.1% final concentration). Cross-linked complexes were analyzed on 13% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and gels were scanned with a Typhoon 9400 Imager (GE Healthcare) and stained with Coomassie blue.
Microfluidics diffusional sizing measurements and quantification
Briefly, 4 μM Mtb RNAP core and 4.8 μM σ^B^ or σ^A^ and 4.8 μM RbpA (when added) were mixed in 40 μl of binding buffer [20 mM HEPES–KOH (pH 8.0), 150 mM KCl, 0.01% BSA, 5% glycerol] incubated at 37°C for 5 min and dialyzed against binding buffer on 0.025 μm MF-Millipore membrane filters (VSWP) for 15 min. A series of dilutions were prepared with RNAP concentrations from 4 μM to 15 nM. Then, −10 ssDNA labeled with fluorescein at the 5′ end was added at 20 nM final concentration and incubated at room temperature for 20 min. Samples were processed using Fluidity One-W (Fluidic Analytics Ltd). The collected gyration radius values, Rh (nm), were normalized and plotted as radius relative change (RRC): RRC = (Rh–Rh0)/Rh0. Rh0 is the gyration radius for DNA alone and Rh is the gyration radius for RNAP-bound DNA. Values from three technical replicates were averaged and fitted using the Grace software (version 5.1.25) with the Hills equation: RFC = k[RNAP]^n^/([RNAP]^n^ + Kd^n^). [RNAP] is the concentration of RNAP holoenzyme, Kd is dissociation constant, and k the amplitude coefficient.
Negative stain EM
Briefly, 200 nM RNAP core was mixed with 600 nM of σ^B^ subunit and 600 nM RbpA, when indicated, in TB. The −10 ssDNA and fork DNA were added to 1 μM and then samples were incubated at 22°C for 60 min. The reaction mixtures were dialyzed in 10 μl drops on 0.025 μm MF-Millipore membrane filters (VSWP) against the TB at 22°C for 1 h. Then, 3 μl of mixture was spotted on Formvar/Carbon copper 200 mesh grids (Electron Microscopy Sciences), glow-discharged for 10 s using PELCO easiGlow (Ted Pella). Grids were stained with uranyl acetate (1% w/v). Images were collected using an 120 kV JEOL 1200 EX II EM equipped with an EMSIS Quemesa 11 megapixel camera with a nominal magnification of ×50 000 and pixel size of 2.86 Å.
Cryo-EM sample preparation
Briefly, 15 μM Mtb RNAP core was mixed with 22.5 μM σ^B^ in 30 μl of 20 mM HEPES–KOH (pH 8.0), 150 mM KCl, 5 mM MgCl_2_, and 2mM DTT and incubated at 37°C for 5 min. To remove glycerol traces, samples were dialyzed in 10 μl drops on 0.025 μm MF-Millipore membrane filters (VSWP) against the same buffer at room temperature for 1 h. Then, 3.75 μl of DNA oligonucleotide was added to 22 μM and incubated at 22°C for 30 min. CHAPSO was added to 8 mM immediately before sample freezing. About 3.5 μl of sample was spotted on Quantifoil Ultra AuFoil R2/2 Au 200 mesh grids that were prepared using Fischione plasma cleaner (NanoClean model 1070). Grids were flash-frozen in liquid ethane using Vitrobot Mark IV (FEI) at 18°C and 90% of humidity.
Cryo-EM data acquisition and processing
Data were collected using a spherical aberration (Cs)-corrected Titan Krios S-FEG instrument (FEI) operating at 300 kV acceleration voltage and equipped with a Gatan K3 Summit direct electron detector (Gatan, Warrendale, PA) and a Gatan BioQuantum energy filter. A total of 9202 movies (50 frames) were collected at an exposure rate of 27.91 e^−^/Å^−2^/s and total electron dose of 55.735 e^−^/Å^−2^ over a nominal defocus range from −0.8 to −2.5 μm, at a nominal magnification of ×81 000 with a physical pixel size of 0.862 Å. Semi-automatic image acquisition was performed with SerialEM [59]. Motion correction, dose weighting, CTF parameter estimation, and particles picking were carried out using WARP [60]. A set of WARP-selected 695 496 particles was used for further processing. Particles with a box size of 360^2^ pixels underwent several 2D classification rounds in cryoSPARC [61]. A cleaned dataset of 371 233 particles was used to compute three ab initio 3D models (three classes). The resulting best ab initio model and two junk 3D models were used as references for the 3D heterogeneous refinement and classification. A cleaned set of 290 345 particles from the best 3D class was used for the final local nonuniform refinement resulting in the consensus I cryo-EM map refined to 3.19 Å (Supplementary Table S2).
3D variability analysis in cryoSPARC
The set of 695 496 particles was re-extracted with box size of 540^2^ pixels and underwent several 2D classification rounds in cryoSPARC. A subset of 394 448 particles from the best 2D classes was used for the ab initio reconstruction to produce five reference volumes: three junk classes, one class corresponding to RNAP dimers, and one class corresponding to RNAP monomers. These volumes were used for the heterogeneous refinement/classification with a larger dataset of 476 952 particles. Next, a subset of 368 449 particles that represented RNAP dimers and monomers was used for the heterogeneous refinement/classification with four reference volumes. The heterogeneous refinement produced two classes of RNAP dimers and one class of RNAP monomers. The final clean dataset of 167 825 particles that represented RNAP monomers was re-extracted with box size of 360^2^ pixels and used in nonuniform refinement to produce the consensus II cryo-EM map refined to 3.33 Å.
Separating clamp conformations
A clean dataset of 167 825 particles that included RNAP monomers was used in 3D variability analysis (3DVA) processing with three principal components (reaction coordinates) and a mask excluding σR4. Particles were sorted in three clusters over coordinate 1. The resulting three maps were used as references to classify the set of 167 825 particles using heterogeneous refinement. Class 0, which represents RNAP with the unswiveled clamp (usRNAP) conformation (36 319 particles), was refined to 3.79 Å. Class 2, which represents RNAP with the swiveled clamp (swRNAP) conformation (21 873 particles), was refined to 4.33 Å.
Separating docked and undocked conformations of σR4
The 3DVA processing was repeated with a mask to select σR4. Particles were sorted in four clusters over coordinate 1. The resulting four volumes were used as references to classify the set of 167 825 particles using heterogeneous refinement. Class 1 comprised docked σR4 (72 799 particles) and was refined to 3.43 Å. Class 2 included undocked σR4 (67 957 particles) and was refined to 3.48 Å.
Model building and refinement
The coordinates of the Mtb Eσ^B^ holoenzyme (PDB ID: 7PP4) were used as starting model. The staring model of full length σ^B^ was built by AlphaFold. The model of the lacUV5 DNA oligonucleotide was built by Coot [62]. The molecular models were assembled and fitted to the cryo-EM map using UCSF Chimera [63] and manually modified with Coot. Fitting of the RNAP clamp and lobe domains was adjusted using rigid body real space refinement in Phenix [64]. Full cycles of real space refinement in Phenix were performed with secondary-structure restrains and geometry optimization. The refined models were manually adjusted in Coot (Supplementary Table S2).
RNAP conformational heterogeneity analysis in cryoSPARC
3DVA analysis was performed in cryoSPARC with the 167 825 particles using three variability components (eigenvectors). For each component pair, particles were separated in nine clusters and the corresponding cryo-EM maps were calculated. The generated 27 maps and the consensus I molecular model were used for rigid body real-space refinement with Phenix. [64]. Models were aligned with Chimera [63] relative to the RNAP α subunits. Distances between atoms where calculated using custom Python scripts. Clamp rotation was measured with PyMOL Molecular Graphics System using the draw_rotation_axis.py script.
Results
The promoter −10 ssDNA triggers large-scale conformational changes in the EσB holoenzyme
Our previous smFRET studies showed that binding of promoter dsDNA to Eσ^B^ does not affect σ^B^ conformation, whereas synthetic upstream fork (us-fork) DNA induces formation of the “open” σ^B^ conformation, like RbpA [50]. Here, using negative stain EM, we observed that the Eσ^B^ octamer disassembled in the presence of us-fork DNA or RbpA (Fig. 1A and B). We hypothesized that the ssDNA segment of the us-fork comprising the promoter −10 element sequence was responsible for the observed RNAP conformational change. Indeed, addition of a synthetic DNA oligonucleotide (−10 ssDNA) that contains the “perfect consensus” −10 element sequence (T_−12_A_−11_T_−10_A_−9_A_−8_T_−7_) to the Eσ^B^ octamer was sufficient to induce its dissociation (Fig. 1B and C). To identify the nature of the RNAP conformational changes induced by −10 ssDNA, we determined the structure of the Eσ^B^/−10 ssDNA complex by single-particle cryo-EM to a nominal resolution of 3.2 Å (consensus I map; Fig. 1D, Supplementary Fig. S1, and Supplementary Table S2). In the published structure of the Eσ^B^ octamer, only 44% of the σ^B^ polypeptide was resolved and the cryo-EM density of the σ^B^ subunit C-terminal domains σ3 and σ4 was missing [51]. Here, the Eσ^B^/−10 ssDNA complex displayed the complete cryo-EM density of the σ^B^ subunit (93% of σ^B^ resolved) with the C-terminal domain σ^3^ contacting the β-protrusion, and the domain σ4 inserted into the RNA exit channel and contacting the β-flap (Fig. 1D and E and Supplementary Fig. S2A). The overall σ^B^ fold was identical to that of the primary σ^A^ subunit in the published structures of RNAP–promoter complexes [32, 47], with a root mean square deviation of 1 Å. The cryo-EM densities of domains σ2 and σ3 were well defined (resolution: 2.9–3.5 Å). Conversely, the densities of σR1.1 and σR4 were poorly resolved (resolution: 4–6 Å) revealing their high conformational mobility. The central part of the RNAP core, including its active site, displayed the highest resolution (between 2.2 and 3.0 Å), while the mobile domains, β′-clamp with bound ssDNA and β-lobe, displayed a lower resolution (between 2.7 and 5 Å) (Supplementary Fig. S1).
We previously showed that the Mtb RNAP core and Mtb Eσ^B^ holoenzyme adopt poorly active conformations, characterized by a wide open clamp and a mobile β-flap [51]. Superposition of the Eσ^B^ and Eσ^B^/−10 ssDNA structures revealed that the RNAP core displayed large-scale rearrangements during holoenzyme maturation induced by the −10 ssDNA (Fig. 2A and B and Table 1). The clamp domain (β′ residues 4–423 and 1219–1261 and β residues 1041–1147), attached to the main RNAP body through switches 1, 2, 3, and 5, rotated 22° orthogonal to the main channel, and adopted a closed conformation (Fig. 2B, red). The β-lobe (residues 180–370, blue) rotated 3.59° toward the clamp, bringing the β subunit gate loop (β-GL) close to the σR1.2 and blocking the access to the active site cleft. The β-flap rotated toward the β′-dock and was fastened by σR4 in a conformation found in all RNAP–promoter complexes structures. In addition, parts of the clamp/jaw/shelf and dock of the β′ subunit (referred to as swivel module [65]) exhibited different extents of rotation parallel to the main channel (Fig. 2C and D). A similar movement, called swiveling, was first observed in the paused elongation complex (PEC) formed by Eco RNAP (Eco PEC) [65, 66] and later in Mtb PECs [67, 68]. The Mtb RNAP active site elements (β′ 423–562) that hold the catalytic Mg^2+^ ion and the bridge helix (BH; β′ 850–882) also underwent conformational changes upon maturation (Fig. 2D insert). Indeed, in the Eσ^B^ octamer, the BH was kinked at β′-A864, adopting the catalysis-inhibited conformation observed in PECs [67]. After σR3–σR4 loading, BH adopted a catalysis-ready conformation. Overall, 78% of the β′ subunit residues and 30% of the β subunit residues underwent conformational changes. We concluded that the Mtb RNAP core and Mtb Eσ^B^ holoenzyme adopt a catalytically inactive swiveled conformation, corresponding to an energetically favored, relaxed state. σR3 and σR4 loading onto the RNAP core, induced by −10 ssDNA, forces RNAP to adopt an unswiveled, catalytically active conformation that is competent to form RPo and initiate RNA synthesis. These results explain the anti-pausing activity of the σR4 in Eco σ^70^ and Mtb σ^B^ and its capacity to stimulate the initial RNA synthesis [69].
Structural transitions in Mtb RNAP during holoenzyme maturation. (A) Cartoon showing the transitions in the Mtb EσB holoenzyme induced by −10 ssDNA binding. (B– D) Structure of the Mtb EσB/−10 ssDNA complex superimposed with the published structure of the Mtb EσB protomer (PDB: 7PP4) from the octameric assembly [51]. The RNAP core is shown as light gray ribbons with cylindrical helices. The σ subunit is omitted. The moving domains of EσB/−10 ssDNA [clamp (β′ 4−419, 1219–1261; β 1117–1140), jaw (β′ 1025–1218), shelf (β′ 882–1011), dock (β′ 444–495), and lobe (β 180–370)] are color-coded as indicated on the left. (C) Ribbon models of the β subunit. On the left, static regions are in gray and the moving domains β-clamp (aa 1041–1147), β-lobe (aa 180–370), and β-flap (aa 808–832) are in red, blue, and khaki, respectively. The switch 3 region (Sw3) is delimited by the Cα atoms of β-G1047 and β-G1065 depicted as spheres. On the right, superposition of the β subunit from the EσB octamer (dark orange) with the β subunit from the EσB/−10 ssDNA complex (slate blue). (D) Ribbon models of the β′ subunit. On the left, static regions are in gray and the moving domains β′-clamp (aa 4–423; 1219–1261) in red, jaw (aa 1025–1218) in orange, shelf (aa 882–1011) in purple, dock (aa 444–495) in firebrick, β′ C-terminus (β′-CT, aa 1262–1283) in cyan, and active site (aa 423–443; 496–562) in magenta. The σ subunit binding domains, β′-CH (aa 339–383) and β′-ZBD (aa 53–85), are indicated. The position of the switch regions is delimited by the Cα atoms of β′-S1219 (Sw1), β′-G419 (Sw2), and β′-P1259 (Sw5) depicted as spheres. On the right, superposition of the β′ subunit from the EσB octamer (dark orange) with the β′ subunit from the EσB/−10 ssDNA complex (slate blue). Insert shows the changes in the RNAP active site and BH (aa 850–882). The catalytic Mg2+ ion is shown in green.
Changes in the ssDNA-binding interface of σB2
The Eσ^B^/−10 ssDNA structure displayed a well-defined cryo-EM density of nucleotides from −13G to −4G with a resolution between 3.0 and 4 Å (Fig. 3A). This structure included the −10 element (positions −12 to −7) and DIS element (positions −6 to −4) (Fig. 3B and C). The remaining nucleotides were not visible, suggesting that they do not form stable contacts with RNAP. Nucleotides from −13 to −5 interacted only with the σ^B^2 domain that encompasses σR1.2 to σR2 (Fig. 3C). The −4G of DIS can interact with the conserved residues β-R282 (Eco β-R371) and β-E285 (Eco β-E374) of β-GL (Supplementary Fig. S2D). In RPo, when the clamp adopts a fully closed conformation, these residues contact the nontemplate DNA strand at positions −4 and −5 [70]. β-GL and β′-CH/σ_2_, direct the nontemplate DNA strand to the main channel and contribute to RPo stabilization [71]. Due to the weak −4G density, we concluded that β-GL does not make stable contacts with ssDNA, but stabilizes binding by sterically restraining the ssDNA 3′-end fluctuations. The overall path of −10 ssDNA and network of interactions between nucleotides and amino acids (Fig. 3B and C) matched that of the nontemplate DNA strand of the transcription bubble in published structures of Eco Eσ^70^ RPo [72] and Mtb Eσ^A^ promoter melting intermediate RPi2 [32] (Supplementary Fig. S3). Thus, we concluded that domain σ2 is a major determinant for the path of the nontemplate DNA strand in RPo.
Architecture of the σB/−10 ssDNA interactions and their quantitative analysis. (A) Cryo-EM density (as mesh) and molecular model of −10 ssDNA. (B) Comparison of the σ/ssDNA binding interfaces in the Mtb EσB/−10 ssDNA complex (on the left) and Taq σA/−10 ssDNA complex [9] (PDB: 3UGP, on the right). The σ domain 2 is shown as a molecular surface colored in wheat. Residues interacting with ssDNA are in orange (σR2) and tomato (σR1.2). The σB-R147 making holoenzyme-specific π-interactions with σB-W144 is colored in light green. ssDNA is shown as a stick molecular model with filled sugars and bases. (C) Schematic drawing showing the network of interactions between ssDNA and the σB subunit in the Mtb EσB/−10 ssDNA complex. Resolved DNA bases are color-coded as in Fig. 1C. Unresolved bases are in white. The amino acid residues interacting with ssDNA are indicated and colored as in panel (B). Lines are colored according to the interaction type (list on the right). (D) Web logo and alignment of σR1.2 and σR2. The ssDNA contacting residues are colored as follows: polar amino acids (G, S, T, Y, C, Q, and N) in green, basic amino acids (K, R, and H) in blue, acidic amino acids (D and E) in red, and hydrophobic amino acids (A, V, L, I, P, W, F, and M) in black. The arrows on the top indicate the contacts observed in the RNAP holoenzyme and missing in the Taq σA/−10 ssDNA complex. (E) Probing of the DNA–protein interactions by formaldehyde cross-linking. Mtb RNAP core (E), σA (A), σB (B), and/or RbpA (as indicated) were cross-linked to fluorescent −10 ssDNA and resolved on SDS–PAGE. (F) Bar graph showing the quantification of the experiment in panel (E). Data are presented as mean values ± standard deviation (SD) of six independent experiments. (G) Measurement of the apparent Kd of −10 ssDNA and the σB subunit by MDS. The relative change RRC of the mean Rh is plotted in function of the protein concentration. The calculated values of the apparent Kd are shown. Measurements were performed once for σB and RbpA–σB, twice for RbpA, three times for EσB, and four times for RbpA–EσB. Data are presented as mean values ± SD.
To understand how the RNAP core stimulates −10 ssDNA binding to σ2, we compared the Eσ^B^/−10 ssDNA structure with the published crystal structure of the Taq σ^A^2–3 fragment in complex with −10 ssDNA [9] (Fig. 3B). The overall path of the −10 element nucleotides (blue) was identical in the two structures. However, the orientations of residues −6 to −4 in DIS (pink) and of the “us-fork region” residues −13 to −12 (gray) were different. In the Mtb Eσ^B^/−10 ssDNA complex, two DIS nucleotides (−5T and −6G) contacted the groove formed by σR1.2 (residues σ^B^-R28, σ^B^-L31) and σR2.1 (σ^B^-R96). In the Taq σ^A^2–3/−10 ssDNA structure, these interactions were missing because DIS nucleotides were displaced outside the σR1.2 groove. The lack of interaction with DIS explains the difference in affinities to −10 ssDNA displayed by the free σ subunit and RNAP holoenzyme reported earlier [16, 19] and see below. The −12T nucleotide of the us-fork region, which is recognized as a base pair in RPo [24, 73], was unpaired in the Mtb Eσ^B^/−10 ssDNA complex analogously to the melting intermediate complex T–RPi1 [12]. In the Eσ^B^/−10 ssDNA complex, −12 T interacted with σ^B^-Q148 (Eco σ^70^-Q437 and Taq σ^A^ -Q260) and with the invariant σ^B^-W144,145 residues of σR2.3 (W-dyad, Eco σ^70^-W433,434 and Taq σ^A^ -W256,257). The W-dyad was stabilized in a “chair-like” conformation through π-stacking with σ^B^-R147 (Fig. 3C and Supplementary Fig. S2D). The W-dyad chair-like conformation is characteristic of RPo [73] (Supplementary Fig. S2F). For Eco Eσ^70^, it has been shown that W-dyad isomerization from the edge-on to the chair-like conformation occurs after nucleation of −11A melting, but before melting of +1, and correlates with the transcription bubble propagation from −11 up to −9 [12]. In T–RPi1, −11A was flipped out, the but W-dyad was still in the edge-on conformation [12] (Supplementary Fig. S2E). In the Taq σ^A^2–3/−10 ssDNA structure, −12T was displaced and could not interact with Taq σ^A^ -Q260 (σ^B^-Q148) and W-dyad [9]. The W-dyad of the Taq σ^A^2–3/−10 ssDNA complex was in the edge-on conformation, like in free σ^70σ^ [74] and in RNAP holoenzymes (Supplementary Fig. S2A–C). Therefore, we suggest that binding of the ssDNA segment between −11 to –5 to σ forces W-dyad isomerization into the chair-like conformation that is further locked by interaction with σ^B^-R147 and stacking with −12T. As we did not observe this RPo-specific conformation in the published Taq σ^A^2–3 apo complex [9], we suggest that β-GL stabilizes the interaction of σR1.2 with DIS and thus in turn stabilizes upstream −10 ssDNA contacts. Functional studies demonstrated that Taq σ^A^-W256 (σ^B^-W144) is not required for −10 ssDNA binding, but is implicated in RPi isomerization to RPo [73]. Therefore, ssDNA binding to domain σ2 acts as a trigger for isomerization in the context of the RNAP holoenzyme. To conclude (i) σ binding to the RNAP core does not change the ssDNA-binding interface of domain σ2; (ii) −10 ssDNA binding to the RNAP holoenzyme leads to changes in the ssDNA-binding interface of domain σ2, such as W-dyad isomerization and contacts formation between σR1.2 and the DIS element; and (iii) contacts with DIS might be stabilized by the interactions with β-GL. These findings explain a large set of biochemical data showing that the RNAP core stimulates −10 ssDNA binding to σ2 [16, 18, 75].
The RNAP core and RbpA stabilize the interaction of the σ2 domain with −10 ssDNA
Eco σ^70^ and Taq σ^A^ models have been used to investigate how −10 ssDNA binding to σR2 is stimulated by the RNAP core [9, 16, 18, 75]. However σ^70^ and σ^A^ differ from Mtb σ^A^/σ^B^ by the presence of large insertions in their NCR. These insertions could partly explain the observed “stimulation”. To determine whether the Mtb RNAP core also stimulates Mtb σ^A^/σ^B^ binding to −10 ssDNA, we used a formaldehyde cross-linking assay that gives a relative estimation of protein–ssDNA affinity [58, 76]. After cross-linking the RNAP core, free σ^B^ subunit and Eσ^B^ to −10 ssDNA (end-labeled with Cy5) in the presence or absence of RbpA, we analyzed the obtained DNA–protein complexes by SDS–PAGE followed by Cy5 fluorescence imaging (Fig. 3E) or Coomassie blue staining (Supplementary Fig. S4A). In agreement with previous findings using Eco Eσ^70^ [16, 18], the Mtb RNAP core produced one slow-migrating cross-linked species with a mobility that corresponded to the β/β′ subunits. Conversely, we observed only a weak cross-linking between ssDNA and the free σ^B^ subunit. In the presence of RbpA, we detected a fast-migrating band at the bottom of the gel, above the free ssDNA that we assigned to RbpA–ssDNA cross-linking (marked as “RbpA–ssDNA”). The σ-specific cross-linking was increased by ∼2-fold in the presence of the Mtb Eσ^B^ holoenzyme compared with free σ^B^ (Fig. 3E and F). This indicated that the Mtb RNAP core stimulated −10 ssDNA binding to the σ^B^ subunit, like the Eco RNAP core to σ^70^. Addition of RbpA to free σ^B^ resulted in a similar level of σ^B^–ssDNA interaction stimulation, as observed with the RNAP holoenzyme alone (∼3-fold higher stimulation relative to free σ), indicating similar affinities of −10 ssDNA for the σ^B^–RbpA complex and the Mtb Eσ^B^ holoenzyme. The combination of σ^B^, RbpA, and Mtb RNAP core resulted in a cooperative effect that was reflected by the further increase of σ-specific cross-linking. We concluded that both RbpA and RNAP core stabilize the interaction of σ^B^2 with the −10 element. Cross-linking experiments with the σ^A^ subunit demonstrated that RbpA also stimulated binding of −10 ssDNA to free σ^A^ and to the Eσ^A^ holoenzyme to a similar extent as observed with σ^B^ (Fig. 3E). RbpA stimulated σ^B^–ssDNA cross-linking even in the presence of a truncated −10 ssDNA (positions −12 to +1) that lacked the RbpA-contacting nucleotides −14/−15 (Supplementary Fig. S4B). Conversely, Hubin et al. [47] reported that RbpA does not affect binding of truncated −10 ssDNA to the σ^A^ subunit in the context of the Mtb RNAP holoenzyme. The origin of this discrepancy remains unclear.
To quantify the interactions between the σ subunit and −10 ssDNA, we used microfluidics diffusional sizing (MDS) to measure the average gyration radius (Rh) of diffusing macromolecules labeled with a fluorescent probe. To decrease possible artifacts from protein aggregation, we used a high ionic strength buffer (150 mM KCl) without Mg^2+^ ions. The Rh value of −10 ssDNA end-labeled with fluorescein was ∼1.9 nm. Addition of Eσ^B^ increased the Rh value to ∼10 nm (Supplementary Fig. S4C). We did not observe any significant change using an oligonucleotide that carried a substitution of the invariant “master” base A_−11_ to C in the −10 motif (−11C ssDNA) to abolish the interaction between σR2 and the −10 element [9, 10, 77] (Supplementary Fig. S4C). Therefore, we concluded that the MDS assay detected specific interactions between −10 ssDNA and RNAP. Next, we measured the affinity for −10 ssDNA of σ^B^ alone, RbpA–σ^B^ complex, Eσ^B^, and RbpA–Eσ^B^ (Fig. 3G). We presented data as the relative change in gyration radius (RRC), calculated as the change in Rh at a given protein concentration divided by the Rh0 of ssDNA alone: RRC=(Rh− Rh0)/Rh0. RbpA alone did not induce any detectable Rh change, indicating that it does not form a stable complex with ssDNA. The σ^B^ subunit displayed low affinity for −10 ssDNA (apparent Kd of 1.4 μM). In the presence of RbpA, σ^B^ affinity for −10 ssDNA increased by ∼11-fold (Kd ∼ 125 nM). The Mtb Eσ^B^ holoenzyme displayed a 27-fold higher affinity for −10 ssDNA (Kd ∼ 52 nM) than the σ^B^ subunit. Addition of RbpA to the Mtb Eσ^B^ holoenzyme further increased the affinity by ∼1.6-fold (Kd ∼ 34 nM), in agreement with the results of the formaldehyde cross-linking assay. Altogether, these experiments showed that binding to σ^B^ of RbpA or Mtb RNAP core stabilizes its interaction with −10 ssDNA.
RNAP in complex with −10 ssDNA adopts multiple conformational states
The consensus I cryo-EM map of the Mtb Eσ^B^/−10 ssDNA complex revealed the conformational mobility of the RNAP clamp and σ^B^R4. To find the range of conformational states adopted by RNAP in our sample, we performed a 3DVA using cryoSPARC [61, 78]. The 2D-aligned images of the Eσ^B^/−10 ssDNA complex revealed two particle populations: RNAP monomers and RNAP dimers (Supplementary Fig. S5, 2D classification). RNAP dimers are likely a predecessors of its octameric state. However, their oligomerization to octamers was blocked by σ^B^R3–σ^B^R4 loaded into the RNA channel. For the 3DVA, we selected a subset of particles that included only RNAP monomers. First, we refined the Eσ^B^/−10 ssDNA monomer map (consensus II map) to a nominal resolution of 3.3 Å (Supplementary Fig. S5). The structure of this consensus II Eσ^B^/−10 ssDNA complex was identical to that of the consensus I map. Next, we performed 3DVA on the consensus II map using three variability components (eigenvectors) (Supplementary Fig. S6). Reconstruction of a series of twenty intermediate maps over each 3DVA component revealed a full range of RNAP conformational states (Supplementary Fig. S6C and Supplementary Movie S1). The first and the last intermediates represented the boundary states with a maximum amplitude in domain movement relative to the “average” consensus structure (Table 1). We observed three conformational change types: (i) swiveling of the clamp core (β′ residues 4–419 and 1215 −1245; Fig. 4A, pink) parallel to the main cleft with simultaneous closing, orthogonal to the main cleft (components 0 and 2); (ii) closing of the clamp head (β′ residues 97–314 and 1162–1245; Fig. 4B, pink), orthogonal to the main cleft (component 0 and 2); and (iii) swinging of the σR4/β-flap module between the β′-dock and β′-ZBD (component 1) (Fig. 4D). In components 0 and 2, clamp movement was accompanied by β-lobe closing (residues 180–370; Fig. 4A and B, blue). Clamp and lobe movements were gradual, while σR4 hopped between boundary states (Supplementary Figs S7 and S8).
Conformational landscape of EσB bound to −10ssDNA. (A) Conformational changes in the RNAP core for the 3DVA component 0: clamp swiveling and closing. The β lobe and β′ subunit clamp domains that underwent conformational changes are colored. (B) Conformational changes in the RNAP core for 3DVA component 2: clamp closing. The β lobe and β′ subunit clamp domains that underwent conformational changes are colored. (C) Comparison of the position of σR1.1 in the unswiveled EσB/−10ss DNA complex and position of the dwDNA in RPo [32]. RNAP is shown as a molecular surface color-coded as in Fig. 1D. The σB subunit is shown as ribbons. The cryo-EM density of σR1.1 is represented as a yellow surface. (D) Conformational changes for the 3DVA component 1: σR4 swinging between the docked and undocked state. RNAP is represented as a molecular surface. The σR4 and RNAP core domains (β′-dock, β′-ZBD, and β-clamp) contacting σR4 are shown as ribbon models. The contacting residues are shown as ball and stick molecular models. The cartoon on the right shows the map of the interactions between σR4 and RNAP core in the undocked and docked σR4 states. Lines are colored according to the interaction type. (D) Superposition of the σB subunit in the docked and undocked conformations with the promoter DNA structure from the Mtb RPc [40] (PDB: 7KIM).
Region 1.1 of σB occupies the dwDNA channel when the clamp is closed and unswiveled
To determine the structures of the most populated boundary states of the clamp, we refined the maps and sorted particles according to the 3DVA results (Supplementary Fig. S8). We computed two Mtb Eσ^B^ structures that corresponded to boundary clamp states: swRNAP and usRNAP. For both states, the −10 ssDNA conformation was identical except its 3′-end nucleotides −4/−5 that fluctuated between β-GL and β-protrusion. The most noticeable change was in the cryo-EM density assigned to σ^B^R1.1 (residues 1–23) and located in the DNA-binding channel (σR1.1; Fig. 4C). Previously, we showed that in the Mtb Eσ^B^ holoenzyme, residues L17–A24 of σ^B^R1.1 were stacked to the clamp head surface, while the cryo-EM density of σ^B^ N-terminus was missing. In usRNAP, the missing density of σ^B^ R1.1 was well defined and occupied the place of the dwDNA (Fig. 4C). We concluded that in the Mtb Eσ^B^ holoenzyme, σ^B^R1.1 stays in the RNAP DNA-binding channel, but adopts multiple conformational states or unfolds. In usRNAP, σ^B^R1.1 movements are restrained by the clamp/lobe and therefore, its density becomes better defined. We propose that transient clamp opening weakens the interactions of σ^B^R1.1 with RNAP and allows dwDNA to displace the σ^B^R1.1 out of the DNA channel, an essential step for the isomerization from the RPi to RPo.
Docking of σR4 in the RNA channel coincides with the β subunit C-terminus restructuring
To determine the structures of the most populated boundary states of σR4, we performed 3DVA with a mask on σR4 and calculated maps from the particles sorted according to component 1 (Supplementary Figs S6 and S8). Particles were distributed equally between two distinct RNAP conformations (43.4% of docked σR4 and 40.5% of undocked σR4). The respective maps were refined to 3.43 and 3.48 Å. In the Mtb Eσ^B^ bearing docked σR4, the overall σ^B^ path matched that of σ^B^ in the consensus I/II structures and that of σ^A^ in the published structures of Mtb RPo [32]. Moreover, σR4 was inserted in the RNA exit channel and its residues σ^B^-I247, L250, D316, Y321, and S323 contacted the β′-dock domain, residue σ^B^-D278 contacted the β′-ZBD, and residues σ^B^-A245 and V246 contacted the β-clamp (Fig. 4D). The structure of Mtb Eσ^B^ with undocked σR4 was different from all published RNAP holoenzyme structures. First, the RNA exit channel was partially occluded by the C-terminal tail (CTT) of the β subunit (residues 1148–1154) that inserts between the β′-ZBD and the β-clamp (Fig. 4D). Second, σR4 was displaced toward the β′-ZBD and residues σ^B^-H252, E249, and A245 made new contacts with β′-ZBD. The contacts of σR4 with the β′-dock and β-clamp, characteristics of the docked σR4 conformation, were lost (Fig. 4D). Superposition of the undocked Mtb Eσ^B^R4 structure with the published structure of Mtb RPc [48, 79] showed that undocked σR4 was incompatible with binding to the −35 element (Fig. 4E) and should hinder RPc formation on −10/−35 class promoters. Conversely, it should not affect RPc formation on extended −10 class promoters because σR3 positioning was unchanged. Our results revealed the high conformational flexibility of σR4 that may help RNAP to adopt a large spectrum of promoter architectures and provides a target for regulation by σR4-binding transcription factors [49, 80]. β-CTT, visible only in the undocked Mtb Eσ^B^ structure, was missing from all published Mycobacterial RNAP holoenzymes structures, but was observed in the PEC bound to the elongation factor NusG [68]. We hypothesize that β-CTT, found only in a subset of bacterial species, may be implicated in the assembly of the mature RNAP holoenzyme and also in promoter escape when σR4 should be ejected from RNA exit channel.
Binding of the −10 promoter element to RNAP mimics RbpA-induced σR3–σR4 loading
To investigate the mechanism by which −10 ssDNA induces a conformational change in Mtb Eσ^B^, we explored the conformational dynamics of σ^B^ by smFRET. We used a σ^B^ subunit stochastically labeled with the DY547P1 and DY647P1 fluorophores in σR2 (position 151) and σR4 (position 292), as described before [50] (Fig. 5A). The distances between labels in free (unbound) σ^B^ and σ^B^ in RPo were ∼50 Å (closed σ^B^ conformation, EPR = 0.83 and E = 0.78) and 83 Å (open σ^B^ conformation, EPR = 0.41 and E = 0.17), respectively [50] (Fig. 5A). The distance between the Cα atoms of the σ^B^ residues 151 and 292 in the Eσ^B^/−10 ssDNA complex (docked σ^B^R4), was 68 Å, which matches the distance in the Mtb RPo (Fig. 5A). The distance in the Eσ^B^/−10 ssDNA complex with undocked σ^B^R4, was intermediate (56.6 Å). Previously, we showed that RbpA forms a stable complex with free σ^B^ [43], but does not affect its conformational dynamics [50]. Addition of −10 ssDNA to free σ^B^ also had little effect on its conformation (∼70% of molecules remained in the closed state) (Fig. 5B), in agreement with the cross-linking experiments (Fig. 3E). Adding both, −10 ssDNA and RbpA resulted in a multimodal distribution, that we approximated by a three-peak Gaussian model with (i) a peak corresponding to the closed conformation of σ^B^ (EPR= 0.87, 22%), (ii) a peak at low FRET efficiency that would correspond to the open conformation of σ^B^ (EPR= 0.31, 34%), and (iii) a third peak with an intermediate FRET efficiency (EPR= 0.63, 44%) (Fig. 5B). We hypothesized that cooperative binding of the −10 ssDNA and RbpA to σ^B^ might increase its dwell time in a partially open conformation but is not sufficient to stabilize the fully open conformation found in the RNAP holoenzyme. This correlated with the increased affinity of σ^B^ to −10 ssDNA (Fig. 3E–G). We suggest that binding of −10 ssDNA to the RbpA–σ^B^ complex is stabilized through direct interactions with both proteins. In the holoenzyme (Mtb Eσ^B^), σ^B^ was mainly found in its closed conformation as reported earlier [50] (Fig. 5C). Addition of −10 ssDNA resulted in a significant increase in the low FRET subpopulation (EPR= 0.31) (Fig. 5C), leading to a pattern that resembled the one observed in the complex of RNAP with us-fork DNA [50]. This suggested that −10 ssDNA binding to domain σ2 stimulates RNAP core-dependent conformational changes in σ^B^ and shifts the equilibrium toward the open σ^B^ conformation with σR4 loaded onto the RNA exit channel. Addition of both −10 ssDNA and RbpA to the Mtb Eσ^B^ holoenzyme shifted the equilibrium toward low FRET (EPR= 0.29) and 20% more molecules adopted the open conformation (Fig. 5C). This change may reflect an increase in the number of molecules that comprise docked σ^B^R4. We concluded that the simultaneous interaction of RbpA with −10 ssDNA and σ-NCR stabilizes −10 ssDNA interaction with σ^B^R2, which in turn prevents the spontaneous collapse of σ^B^ to the closed conformation and favors σ^B^R3–R4 loading to RNAP.
Conformational changes in σB upon the −10 ssDNA and RbpA binding. (A) Molecular models of the free σB subunit (ribbons with cylindrical helices) and EσB in the undocked and docked conformations. The RNAP core is depicted as a molecular surface. Spheres represent the Cα atoms of the σB subunit residues Cys-151 and Cys-292 (color-coded as indicated on the left) labeled with the DY-547P1 and DY-647P1 fluorescent dyes. Distances between dyes, shown above the models, were calculated using the smFRET data (in blue) [50] and from the EσB structures reported here (in black). (B) smFRET of free σB performed without ligands or in the presence of −10 ssDNA (DNA) and RbpA. (C) smFRET of the Mtb EσB holoenzyme in the presence of −10 ssDNA, −11C ssDNA, wild-type RbpA, and the RbpA double mutant R88,89A (RbpAmut). The table lists the EPR values (E), percentages (P) of molecules in each histogram peak, and total number (N) of molecules from panels (B) and (C). ND, not determined. All smFRET measurements were done three times except for EσB and RbpA–EσB which were done twice.
Interaction of σ2 with −10 ssDNA and RbpA σ-binding domain induces σR3–σR4 loading
To verify that the smFRET efficiency change induced by −10 ssDNA was due to its sequence-specific interaction with σR2, we performed experiments with −11C ssDNA (Fig. 5C). Addition of −11C ssDNA to the Mtb Eσ^B^ holoenzyme did not induce any increase in the low FRET population, suggesting that σ^B^ remained in the closed confirmation. The EPR distribution pattern matched that of the RNAP holoenzyme. Addition of RbpA and of −11C ssDNA to the Mtb Eσ^B^ holoenzyme resulted in the increase of the low FRET subpopulation (EPR= 0.28, 60%), as observed with the RbpA–Eσ^B^/−10 ssDNA complex (EPR= 0.31, 53%) (Fig. 5C). This is likely due to stabilization of the σ^B^/−10 ssDNA interaction by RbpA that compensates for the defect introduced by the −11C mutation. Interaction of RbpA with the σ^B^ subunit NCR (σ^B^-NCR) is essential for transcription activation [47, 48]. To verify that the observed effect of RbpA on σ^B^ conformation was due to its interaction with σ^B^-NCR, we performed smFRET experiments with a mutant RbpA harboring R88A and R89A in its σ-binding domain (Fig. 5C, panels RbpA^mut^). These alanine substitutions abolish RbpA–σ interactions and RbpA-mediated transcription activation [42]. As expected, the two mutations suppressed σ opening by RbpA. Addition of −10 ssDNA to the RbpA^mut^–Eσ^B^ complex produced the same pattern as −10 ssDNA alone, showing the stimulating effect of RbpA loss. Finally, combining mutant −11C ssDNA with RbpA^mut^ resulted in an EPR distribution similar to that of the Eσ^B^ holoenzyme. Based on these results, we concluded that σ loading is induced by the specific interaction of RbpA and −10 ssDNA with their respective binding sites on the RNAP holoenzyme and that RbpA and −10 ssDNA are interchangeable (Fig. 5C, bottom).
The β subunit flap is essential for RbpA-driven σR3–σR4 loading
Binding of σR4.2 to the β subunit flap-tip-helix (β-FT) domain (Fig. 6A) is essential for positioning σR4.2 to interact with the −35 sequence element [81], but not for the formation of the Mtb Eσ^B^ immature holoenzyme [51]. To determine whether the β-FT/σR4.2 contact was essential for RbpA and −10 ssDNA-mediated σR3–σR4 loading, we performed transcription assays and smFRET measurements with the mutant Mtb E^ΔFT^ in which the β subunit residues 811–825 were deleted. The mutant Mtb E^ΔFT^ core is active in the promoter-independent initial transcription assays with a DNA scaffold template [69]. Here, we first tested the activity of the Mtb E^ΔFT^σ^B^ holoenzyme in transcription runoff assays with the RbpA-dependent sigAP promoter that contains an almost perfect −10 sequence element (five of six matches) and displays weak homology to the −35 sequence element (three of six matches) (Fig. 6B and C) [44]. The β-FT deletion almost fully suppressed the transcription stimulation by RbpA, suggesting that RbpA functioning strongly depends on the β-flap. Previous studies on Eco RNAP showed that β-FT is dispensable for transcription initiation from the extended −10 class promoters without the −35 motif. In this context, interaction of σR3 with the T_−17_R_−16_T_−15_G_−14_ motif of the extended −10 element is essential for recognition of the extended −10 class promoters, while interaction of σ4.2 with the −35 motif is dispensable [82]. The extended −10 motif stimulates transcription initiation by Eσ^B^ independently of RbpA [46]. When we tested the activity of the mutant Mtb E^ΔFT^σ^B^ holoenzyme on a synthetic sigAP promoter variant that contained the T_−17_R_−16_T_−15_G_−14_ motif (sigAP-TGTG; Fig. 6B and C), we found that the β-FT of Mtb RNAP was essential for transcription from the sigAP-TGTG promoter (Fig. 6B and C), unlike what observed for Eco RNAP [81]. Addition of RbpA restored the Mtb E^ΔFT^σ^B^ holoenzyme activity to the level of wild-type RNAP. Our results indicate that despite the high level of conservation in the core RNAP structure and function, the Eco paradigm might not be applicable to other bacterial species. The effect of the β-FT deletion was reminiscent of the effect of the deletion of its partner σR4.2 that leads to transcription inhibition from sigAP-TGTG [46].
The RNAP β-flap is essential for σ loading induced by RbpA and by −10 ssDNA. (A) Cartoon showing the σB 4/β-FT interactions. Molecular model of Mtb EσB/−10 ssDNA in complex with RbpA (RbpA coordinates taken from PDB: 6EDT [32]). The RNAP core is depicted as molecular surface. The σB subunit, RbpA, and β-FT are shown as ribbons models. (B) Effect of β-FT deletion on run-off transcription from the sigAP and extended −10 type sigAP-TGTG promoters. The difference in promoter sequences is shown on the top. The σB subunit regions that interact with promoter motifs are depicted as rectangles above the DNA sequences. (C) Bar graph showing the quantification of the run-off RNA products of the gel shown in panel (B). Activity was calculated by dividing the run-off RNA signal (counts) in each lane by the total counts across all lanes. Data are presented as mean values ± SD of three independent experiments. (D) smFRET analysis of the mutant Mtb EΔFTσB holoenzyme (lacking the RNAP β-FT) in the presence of −10 ssDNA and RbpA. The table lists the EPR values (E), percentages (P) of molecules in each peak, and total number (N) of molecules in panel (D). ND, not determined. The smFRET measurements were done three times.
In agreement with the transcription assay findings, smFRET experiments showed that binding of RbpA to the mutant Mtb E^ΔFT^σ^B^ holoenzyme had little effect on EPR distribution (Fig. 6D), compared with RbpA binding to wild-type Mtb Eσ^B^ (Fig. 5C). Only a minor subpopulation of molecules (24% versus 64% for wild type) were in the low-FRET state, corresponding to the open σ^B^ conformation (EPR = 0.34). The distribution of FRET states was similar to that of the Mtb Eσ^B^ holoenzyme in the absence of RbpA (Fig. 5C). We concluded that in the Mtb E^ΔFT^σ^B^ holoenzyme, most σ^B^ molecules remained in the closed conformation. Therefore, when σ^B^R4.2/β-FT interaction is disrupted, RbpA cannot induce the σ^B^ conformational change essential for RPo formation. These results perfectly fit with the mechanism where RbpA promotes the formation of the contacts between σR4.2 and β-FT that are essential for the correct σR3 positioning relative to the extended −10 motif.
The β subunit flap is essential for −10 ssDNA driven σR3–σR4 loading
Then, we tested the effect of β-FT deletion on −10 ssDNA-induced loading of σR3–σR4 to RNAP (Fig. 6D). The addition of −10 ssDNA to Mtb E^ΔFT^σ^B^ resulted in a broad EPR distribution of the molecules, which we fitted with a three-peak Gaussian model. This model indicates that a minority of σ^B^ molecules adopt the fully open state (EPR= 0.28, 14%), while the majority are found in a partially opened (EPR= 0.55, 45%) and closed (EPR = 0.85, 41%) conformations (Fig. 6D, “panel ΔFTσ^B^/−10 ssDNA”). Thus, similar to what we observed for RbpA, β-FT is required for the −10 ssDNA-induced conformational change in the σ subunit. Upon addition of both RbpA and −10 ssDNA (Fig. 6D, panel “RbpA-ΔFTσ^B^/−10 ssDNA”), the equilibrium was further shifted toward the intermediate σ^B^ conformation (EPR= 0.55, 53%). These results suggest that both RbpA and −10 ssDNA bind to the mutant Mtb E^ΔFT^σ^B^, but cannot promote the correct positioning of σR4 due to the absence of β-FT, its anchoring point. Furthermore, RbpA and −10 ssDNA remodel the σ subunit through a similar mechanism by targeting σ2. The effect of −10 ssDNA on σ conformation in the absence of the σR4/β-FT contact on the RNAP core suggests that σ2 is exposed for binding to ssDNA. We propose that ssDNA binding to σ2 destabilizes the overall fold of the σ subunit, which allows σR3 and σR4 to interact with their respective binding sites on the RNAP core.
Clamp movements and σR4 swinging do not correlate
To determine whether there is a correlation between σR4 docking and clamp closure/swiveling, we performed pairwise cluster analysis (nine clusters) on variability components generated by 3DVA with the full consensus II map (Supplementary Fig. S6). We compared the variability components 1 (C1, σR4 swinging) with the variability components 0 (C0, clamp swiveling) and 2 (C2, clamp closing) and also C0 with C2. For each component pair, we sorted particles in nine clusters (Fig. 7A–C and Supplementary Fig. S6C). We used the resulting series of cryo-EM maps for rigid body refinement of the consensus I molecular model. Clamp rotation angle measurement has been widely used to characterize RPo conformational dynamics [32, 35, 83]. However, results depend on the choice of the reference structure. Here, to characterize RNAP domain movements, we measured the distances between reference points in each of the refined models that provide absolute characteristics for a given structure independently of the reference choice (Supplementary Table S1). To quantify the σR4 domain movement, we measured the distances between the Cα atoms of σ-I247 in σR4 and β′-I469 in β′ dock. To quantify clamp/lobe movements, we measured the distances between β-G284 in the β-lobe and β′-K123 in the β′-clamp head and β′-L360 in the clamp core (Fig. 7D). By plotting the distance ranges between σ-I247 and β′-I469 against the distances between β-G284 and clamp (β′-K123 and β′-L360) (Fig. 7A and B and Supplementary Table S1), we found that σR4 adopted two distinct conformations with average distances of ∼7 Å (docked σR4) and ∼17 Å (undocked σR4). We did not detect any intermediate state. This suggests that such states are short-lived. Moreover, we did not find any correlation between clamp and σR4 movements. Unlike σR4, clamp movements were gradual and displayed a clear correlation between swiveling and opening/closing (Fig. 7C and Supplementary Table S1): the open clamp state adopted a more swiveled conformation and the closed clamp state an unswiveled conformation. We concluded that after σR3 binding to the β-lobe and σR4 insertion into the RNA exit channel, the clamp adopts preferentially an unswiveled/closed conformation, but remains dynamic. This is essential for DNA entry into the active site cleft during the isomerization from RPc to RPo.
Correlation between σ loading, σR4 swinging, and clamp motions. (A–C) Graphs showing the distances between σR4 and β′-dock and β′CH and β-lobe in the nine clusters produced in the 3DVA (see Supplementary Table S1). (D) Molecular models showing the above mentioned regions; the Cα atoms of the residues used for the distance calculations are shown as spheres. (E) Box plots showing the compilation of the clamp-lobe distances from published cryo-EM structures of RNAP plotted in function of the transcription cycle progression (reaction coordinates). Dotted lines show the mean values for E. coli and M. tuberculosis (Myco), respectively. N, number of data points used for each reaction step (see Supplementary Table S2). (F) Model depicting the changes in RNAP clamp (light salmon) and σ subunit (σR1.1 in white, color-codes for σR2, σR3, and σR4 as in Fig. 1E) conformations throughout the transcription cycle. Promoter DNA is depicted as a blue ladder, RNA as an orange line. Each complex type (E, Eσ, RPc, RPi, RPo, and EC) and the reaction direction are indicated by arrows at the bottom.
Clamp dynamics gradually decrease during progression of the transcription cycle
To relate the observed β′-clamp/β-lobe movements in the RNAP/−10 ssDNA complex with RNAP conformational dynamics throughout the transcription cycle, we analyzed the β′-clamp/β-lobe states in 40 published cryo-EM structures of bacterial RNAPs, alone and in complex with various transcription factors (Fig. 7E and Supplementary Table S2). The ranges of distances from the β-lobe (β-G284) to the β′-clamp head (β′-K123) and to the clamp core (β′-L360) are presented as box plots for each transcription cycle step: assembly of the core (E) and σ subunit into the RNAP holoenzyme (Eσ), promoter complex formation (RPc, RPi, and RPo), and elongation (EC). The distance analysis showed that the RNAP core adopted the largest open clamp state with a wide range of distances (amplitude ∼10 Å). After RNAP holoenzyme assembly, the clamp displayed a broader range of states (amplitude ∼17 Å), from wide open (matching the RNAP core state) to closed (matching the RPo state). The immature Mtb Es^B^ holoenzyme, in which domains σR3–σR4 are unbound from their respective sites in the core, adopted a wide open clamp state (distances of 32.7/50.6 Å), similar to the RNAP core [51, 84]. We concluded that binding of the σR3 domain to the RNAP β-protrusion and insertion of the σR4 domain in the RNA channel increase the conformational mobility of the core enzyme and promote clamp closure. RNAP binding to promoter DNA duplexes restrained clamp movements in RPc while keeping the wide range of states characteristics of holoenzymes. Nucleation of promoter DNA melting in RPi led to a decrease in clamp mobility (amplitude ∼5 Å); however, the clamp could still adopt partially open states. Upon RPo formation, replacement of the σR1.1 in the DNA channel by the dwDNA froze the clamp in the closed state and limited its conformational dynamics (amplitude ∼2 Å) (Fig. 7E and F). After the promoter escape to elongation, σR3–σR4 are replaced by nascent RNA that favors the σ subunit dissociation. Overall, clamp dynamics increased, but the clamp remained mainly closed over the DNA template. We observed the most closed clamp state in Eco his paused TEC (PDB: 6ASX [65]) and the most unswiveled clamp state in Eco elemental paused TEC (PDB: 8EGB [85]). Although the general tendency was toward a decrease in clamp-lobe distances over the reaction coordinates, we observed some differences in distance distribution in every dataset due to uncoupling between clamp closing and clamp swiveling (most noticeable for RPo and EC). Plotting separately the average distances of the Eco (red dotted line) and Mtb (orange dotted line) datasets (Fig. 7E) highlighted a large data divergence particularly for the RNAP core (E) and RPc. Yet, due to the small number of available structures, this difference was not significant. The clamp-lobe distance range of the Mtb Eσ^B^/−10 ssDNA complex (17.4/38.6 Å, amplitude of ∼5 Å) positioned it between RPi and RPo (Fig. 7E, blue dashed line, R/−10). We suggest that RNAP conformational state in this complex may correspond to that in the transient intermediate on the path to RPo. Unlike in RPi and the Mtb Eσ^B^/−10 ssDNA complex, in RPo, σR1.1 is displaced by dwDNA and the downstream part of the transcription bubble (dw-fork DNA) that enters the DNA channel. Thus, we concluded that binding of dw-fork DNA locks the clamp in the closed state, which is retained during elongation (EC) (Fig. 7F). The results of our analysis are in perfect agreement with the smFRET data showing a wide open clamp in the RNAP core, a partially closed clamp in the RNAP holoenzyme, and a fully closed/locked clamp in RPo [30].
Discussion
The principal finding of our work is that the σ subunit functions as a sensor of regulatory signals from the ssDNA effector, leading to isomerization between the open and closed RNAP clamp conformations. Therefore, as proposed by Ishihama [86] several decades ago, σ is not just a promoter-recognition-melting factor, but also a major regulator of RNAP conformational dynamics. We suggest that in solution, the default “relaxed” conformations of the clamp in the RNAP core and of the σ subunit are open/swiveled and closed with masked DNA binding regions, respectively. Bringing together the two partners leads to σ opening and RNAP clamp closing/unswiveling and consequently to the formation of a “stressed” RNAP, competent to initiate transcription on promoter DNA.
The σ-mediated maturation of the RNAP active site and analogy with NusG/RfaH
The swiveled clamp state was first described as an attribute of the PECs [65] regulated by the universal transcription factor NusG/Spt5 and its paralog RfaH [67, 68, 87]. The anti-pausing Eco NusG suppresses swiveling by contacting β-GL [67]. We showed that σ subunit domains loading onto the nucleic acid binding channels of RNAP induced a catalytically proficient unswiveled conformation of the active site. This finding provides a structural rational for the previously reported anti-pausing activity of Eco σ^70^ and Mtb σ^B^ during initial transcription [69] and reveals a remarkable functional similarity between σ factors and elongation factors that act via universal RNAP domains: β′-CH, β-protrusion, β-flap, and β-GL. We propose that σ-mediated RNAP maturation, conserved between Eco and Mtb, has evolved to suppress nonspecific transcription initiation by the RNAP core and to promote sequence-specific transcription initiation by the RNAP holoenzyme.
Linking σ loading and clamp closure
The clamp closure/unswiveling observed in the Eσ^B^/−10ssDNA complex can be induced by loading of σ domains or/and by −10 ssDNA binding. As −10 ssDNA does not make stable contacts with the RNAP core, we speculate that loading of σR3–σR4, and probably of σ1.1, promotes clamp closure. This conclusion is supported by the comparison of the published cryo-EM structures of RNAPs. (i) The clamp is open in the Mtb RNAP core and RNAP holoenzyme that comprises σ subunits with unloaded σR3–σR4. (ii) Similarly, in the published structures of Mycobacterium smegmatis RNAP core and holoenzyme with unloaded σ^A^R4, the clamp is in the open conformation [88]. (iii) In the RbpA–Eσ^A^ complex [35], which contains fully loaded σR3–σR4, the clamp adopts a more closed conformation than in the RNAP core. (4) In Eco, the clamp is open in the RNAP core and closed in the Eσ^70^ holoenzyme that contains fully loaded σR3–σR4 [84, 89]. Yet, the smFRET findings suggested that the clamp adopts mainly an open state in both Eco RNAP core and holoenzyme, unlike the closed state in RPo [27]. This discrepancy might reflect the fact that the published structures represent a “consensus” conformation of RNAP derived from a conformational ensemble. Indeed, the clamp high conformational mobility, modulated by the transcription factor TraR, has been observed in the Eco Eσ^70^ holoenzyme [83]. We interpret these findings as the evidence of a larger amplitude of clamp motions in the holoenzyme induced by σ loading. In Mtb RNAP, the equilibrium between clamp states is shifted toward the open conformation due to the weak binding of σR3–σR4. In Eco RNAP, the equilibrium is shifted toward the closed conformation due to the stable binding of σR3–σR4.
Stabilization of the closed clamp state by the σ/ssDNA interaction and path to RPo
The presence of a promoter DNA template drastically reduces the clamp and σ domain conformational mobility in RPi/RPo [27, 28]. In the Eσ^B^/−10 ssDNA complex, the clamp adopted a more closed state than in the Mtb RNAP holoenzymes, but more open than in RPo. We infer that ssDNA binding to RNAP restricts the clamp dynamics and shifts its conformational equilibrium toward a closed state, similar to that observed in RPi. Indeed, all early RPis with the melted −10 element show a partially closed clamp state [12, 32, 72]. In RPo, after the replacement of σR1.1 by dwDNA, the clamp becomes locked in the closed state and remains locked during elongation [30]. This scheme fits well with the Mtb model, but may differ from that of Eco (Fig. 7E, dotted lines). Moreover, the conformational equilibrium between clamp states can be influenced by the experimental conditions, such as type of promoter DNA template, divalent ion concentration, and presence of transcription factors (e.g. Eco TraR) [30, 83].
Difference in RNAP holoenzyme assembly pathway in E. coli and M. tuberculosis
Despite decades of studies, the mechanism of RNAP holoenzyme assembly remains poorly understood, and the exact order of events of the σ subunit loading to the RNAP core is unknown [86, 90, 91]. Our results suggest that assembly starts after σR2 binding to β′-CH on the clamp and σR1.1 insertion into the dwDNA channel. The open clamp conformation of the RNAP core and β-flap wobbling allow the entry of the C-terminal domains of σ into RNAP main cleft. Binding of σR3 to the β-protrusion ties the RNAP pincers together and favors clamp closure. Finally, σR4 inserts into the RNA exit channel and binds to the β-flap and β-clamp, stabilizing the whole system. On the basis of the high conservation of the core binding regions of group I/II σ subunits and their respective binding sites on RNAP, the assembly pathway should be universal for all bacteria, but may differ in the nature of bottleneck steps delimited by lineage-specific insertions/deletions in σ and RNAP. For example, in Eco, σ^70^ loading onto the RNAP core is a spontaneous process that does not require any additional co-factor. Conversely, in Mtb, σ^A^ and σ^B^ loading onto the RNAP core is RbpA-dependent. This RbpA dependency is a feature of the Mtb RNAP core and not of the σ subunit because assembly of a chimeric holoenzyme (Mtb σ^B^ and Eco RNAP core) proceeds without RbpA [50].
Allosteric switch in σ and stimulation of the nontemplate DNA strand binding by RNAP core
A large number of biochemical studies on Eco and Taq models reported a core-induced conformational change in the group I σ subunit leading to unmasking of its DNA binding regions and to stimulation of −10 ssDNA binding [9, 73, 92]. Our smFRET and structural studies in Mtb and Eco suggest that the only detectable conformation change in σ is the movement of σR3–σR4 away from σR2, providing a time window for ssDNA binding. The RNAP core stabilizes −10 ssDNA/σ2 interactions by favoring contacts between DIS and σR1.2, thus preventing σ closing. In Mtb, RbpA tethers −10ssDNA to σR2 and prevents σ closing. Similarly, it has been shown that a DNA aptamer containing −10 ssDNA induces Taq σ^A^ opening [93]. However, a σ^70^ fragment that lacks σR3–σR4 (σ2) displayed enhanced binding to −10 ssDNA in the presence of the RNAP core [16] or the β′-clamp fragment alone [20]. Our analysis of cryo-EM structures suggests that the Mtb RNAP core alone does not induce any global conformational change in σ2. Conversely, −10 ssDNA induces W-dyad isomerization that may stabilize contacts between DIS and σR1.2. Although the overall σ2 fold is conserved in all group I–II σ subunits, the regulation of −10 element binding may vary among species due to lineage-specific insertions in σ-NCR. For instance, insertions in the Eco σ^70^ and Taq σ^A^ σ-NCR would clash with β′-CH and impose a conformational change in σR2 upon holoenzyme assembly. A plausible hypothesis is that σR1.2 N-terminal α-helix (Mtb σ^B^ residues 26–36), which is attached to the main body of σR2 through an unstructured linker, may be displaced in free σ and will not form optimal contacts with DIS. Binding to the RNAP core may lock the σR1.2 α-helix in the correct conformation that is optimal for DIS binding.
Multiple RbpA roles in transcription initiation
RbpA stabilizes the RNAP holoenzyme [44], stimulates promoter DNA melting [43, 44], and slows down promoter escape [48, 94]. Our results explain the multiple roles of RbpA in transcription initiation and suggest that RbpA acts from RNAP assembly to promoter escape by stabilizing the σ2/−10 element, σ3/extended −10 element, and σ4/−35 element interactions. Specifically, RbpA induces the optimal fit of RNAP to −10/−35 elements upon RPc formation. Then, RbpA-mediated anchoring of σR2 to −10 ssDNA should stimulate formation of the transcription bubble during isomerization from RPc to RPo. The latter implies that RbpA should slow down promoter escape by inhibiting the disruption of σR2/−10 contacts, in agreement with the kinetics studies [94].
Supplementary Material
gkaf272_Supplemental_Files
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Burgess RR, Travers AA, Dunn JJ et al. Factor stimulating transcription by RNA polymerase. Nature. 1969; 221:43–6.10.1038/221043 a 0.4882047 · doi ↗ · pubmed ↗
- 2Feklistov A, Sharon BD, Darst Sa et al. Bacterial sigma factors: a historical, structural, and genomic perspective. Annu Rev Microbiol. 2014; 68:357–76.10.1146/annurev-micro-092412-155737.25002089 · doi ↗ · pubmed ↗
- 3Gruber TM, Gross CA Multiple sigma subunits and the partitioning of bacterial transcription space. Annu Rev Microbiol. 2003; 57:441–66.10.1146/annurev.micro.57.030502.090913.14527287 · doi ↗ · pubmed ↗
- 4Rodrigue S, Provvedi R, Jacques PÉ et al. The σ factors of Mycobacterium tuberculosis. FEMS Microbiol Rev. 2006; 30:926–41.10.1111/j.1574-6976.2006.00040.x.17064287 · doi ↗ · pubmed ↗
- 5Buc H, Mc Clure WR Kinetics of open complex formation between Escherichia coli RNA polymerase and the lac UV 5 promoter. Evidence for a sequential mechanism involving three steps. Biochemistry. 1985; 24:2712–23.10.1021/bi 00332 a 018.3896304 · doi ↗ · pubmed ↗
- 6Roe JH, Burgess RR, Record MT Kinetics and mechanism of the interaction of Escherichia coli RNA polymerase with the lambda PR promoter. J Mol Biol. 1984; 176:495–522.10.1016/0022-2836(84)90174-8.6235375 · doi ↗ · pubmed ↗
- 7Sclavi B, Zaychikov E, Rogozina A et al. Real-time characterization of intermediates in the pathway to open complex formation by Escherichia coli RNA polymerase at the T 7A 1 promoter. Proc Natl Acad Sci USA. 2005; 102:4706–11.10.1073/pnas.0408218102.15738402 PMC 555702 · doi ↗ · pubmed ↗
- 8Li L, Molodtsov V, Lin W et al. RNA extension drives a stepwise displacement of an initiation-factor structural module in initial transcription. Proc Natl Acad Sci USA. 2020; 117:5801–9.10.1073/pnas.1920747117.32127479 PMC 7084136 · doi ↗ · pubmed ↗