Discovery of Highly Potent BET Inhibitors based on a Tractable Tricyclic Scaffold
Jaffer M. Zaidi, Eleonora Comeo, Andrew Baxter, Alex G. S. Preston, Weng C. Chan, Michael J. Stocks

TL;DR
This paper introduces a new class of potent BET inhibitors based on a tricyclic scaffold that effectively targets BRD4, a protein involved in gene regulation and cancer.
Contribution
The novel contribution is the development of a triazinoindole scaffold with chemomimetic substituents that inhibit BRD4 with high potency.
Findings
The triazinoindole scaffold effectively inhibits BRD4 with low nanomolar affinity.
The lead compound has favorable physicochemical and in vitro stability properties.
Dimethylisoxazole and dimethyltriazole substituents mimic N-acetylated lysine residues in histone tails.
Abstract
The bromodomain and extra-terminal domain (BET) protein family is a class of epigenetic reader proteins that recognize N-acetylated lysine residues in histone tails, playing a crucial role in gene expression and cell transcription. Selective inhibition of bromodomain-containing proteins (BRDs) disrupts transcription in key oncogenes. Over the past decade there has been considerable interest in developing small molecule BET inhibitors for the treatment of hematological malignancies and solid tumors. Herein, we report the development of a triazinoindole scaffold capable of the inhibition of bromodomain-containing protein 4 (BRD4), with either dimethylisoxazole or dimethyltriazole substituents acting as chemomimetics of the N-acetylated lysine residues. Derivatization of the parent scaffold afforded the lead compound, which displays low nanomolar affinity toward BRD4-BD1 with a favorable…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
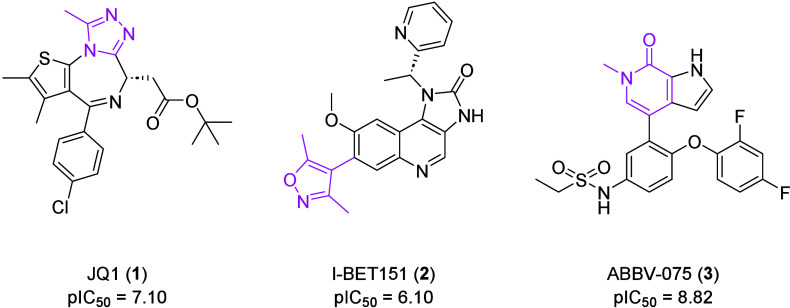 Figure 1
Figure 1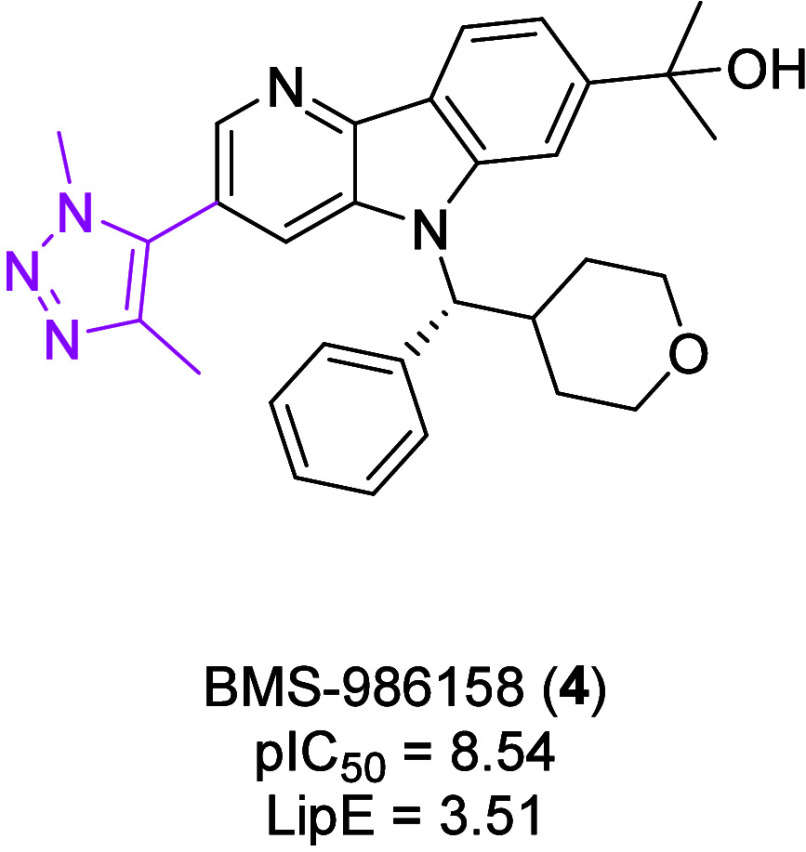 Figure 2
Figure 2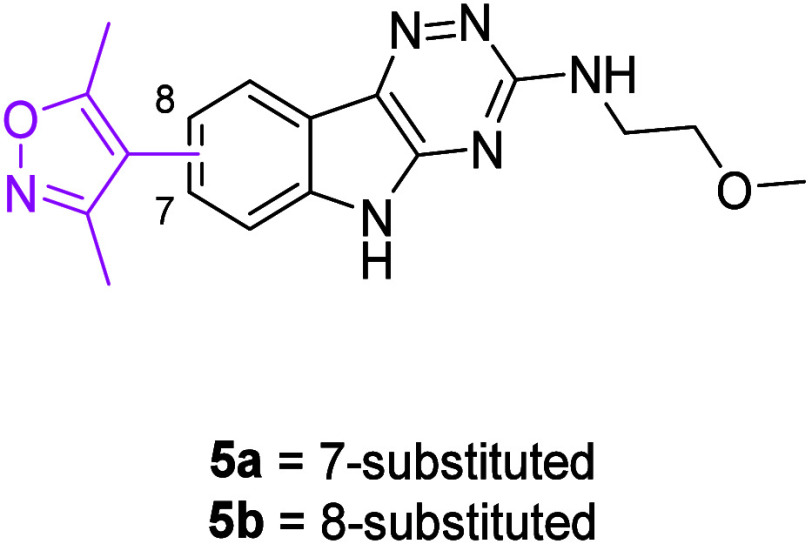 Figure 3
Figure 3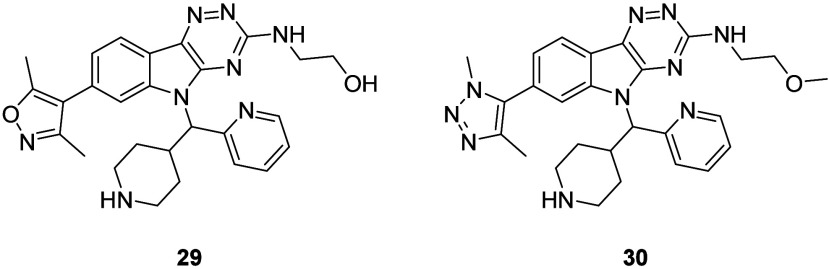 Figure 4
Figure 4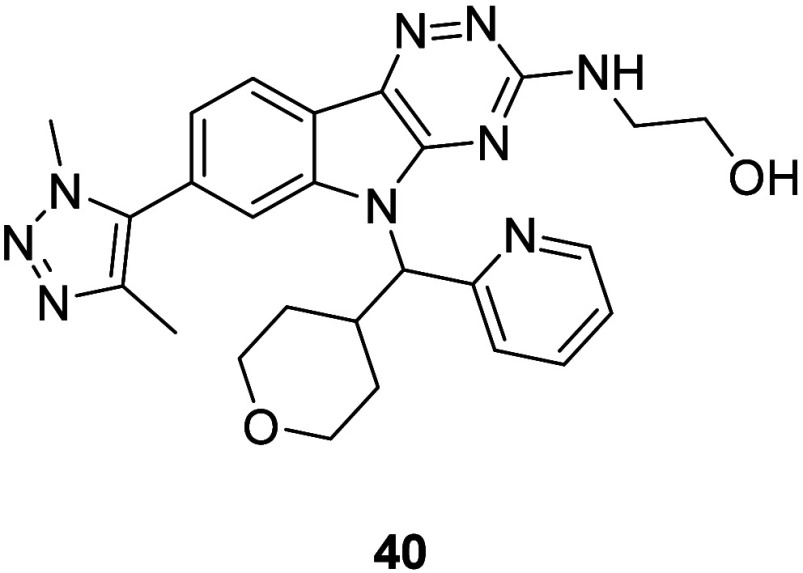 Figure 5
Figure 5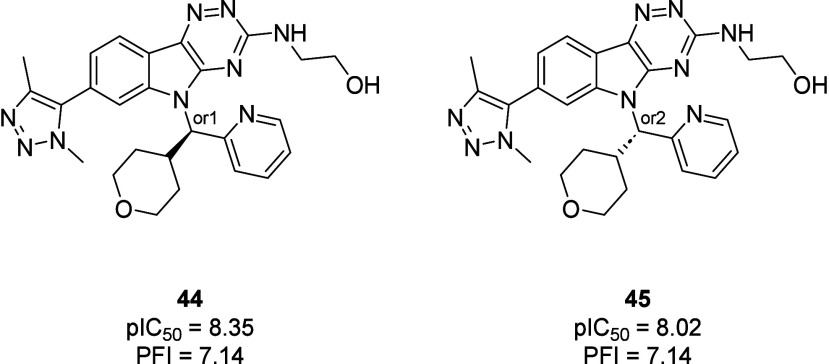 Figure 6
Figure 6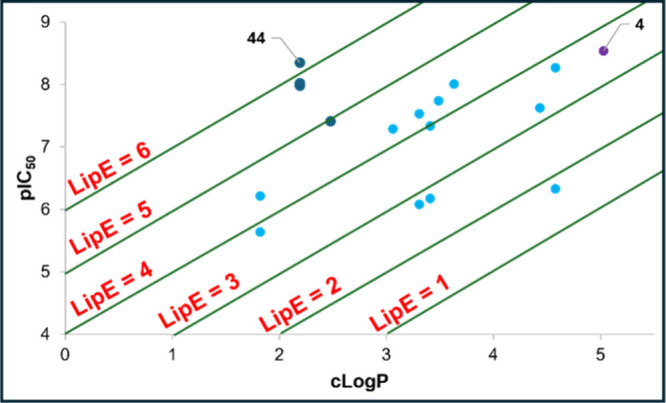 Figure 7
Figure 7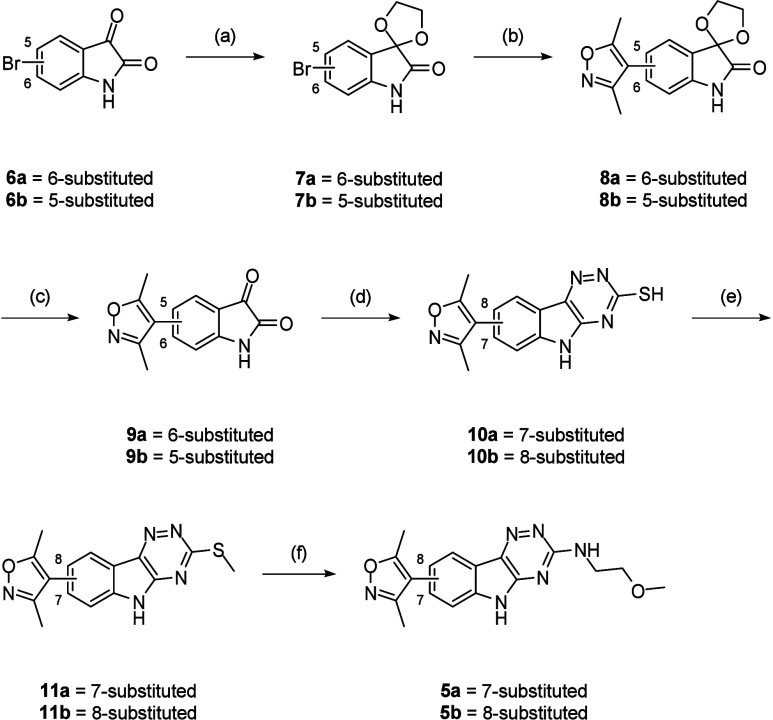 Figure 8
Figure 8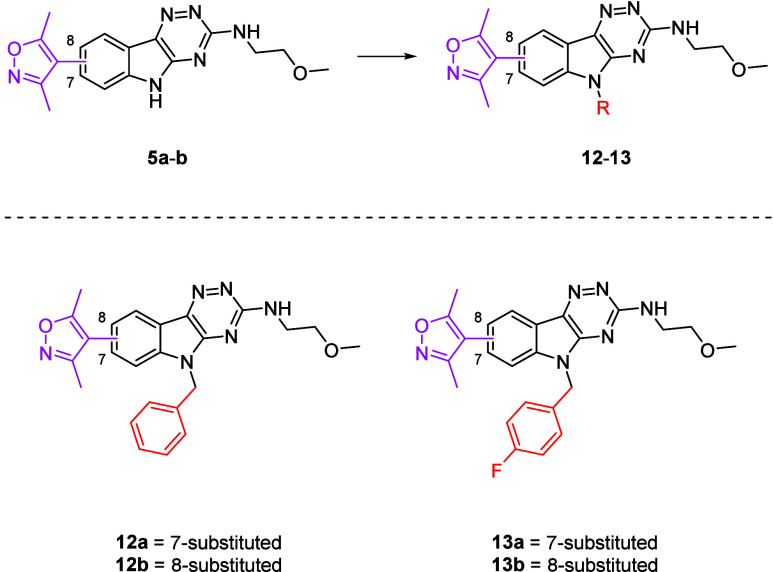 Figure 9
Figure 9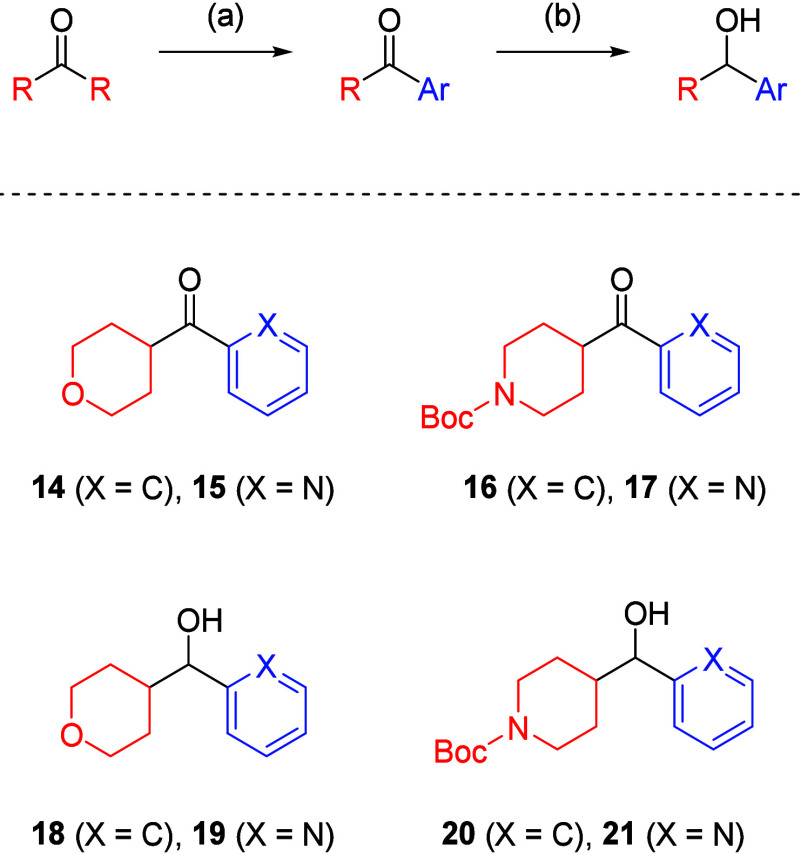 Figure 10
Figure 10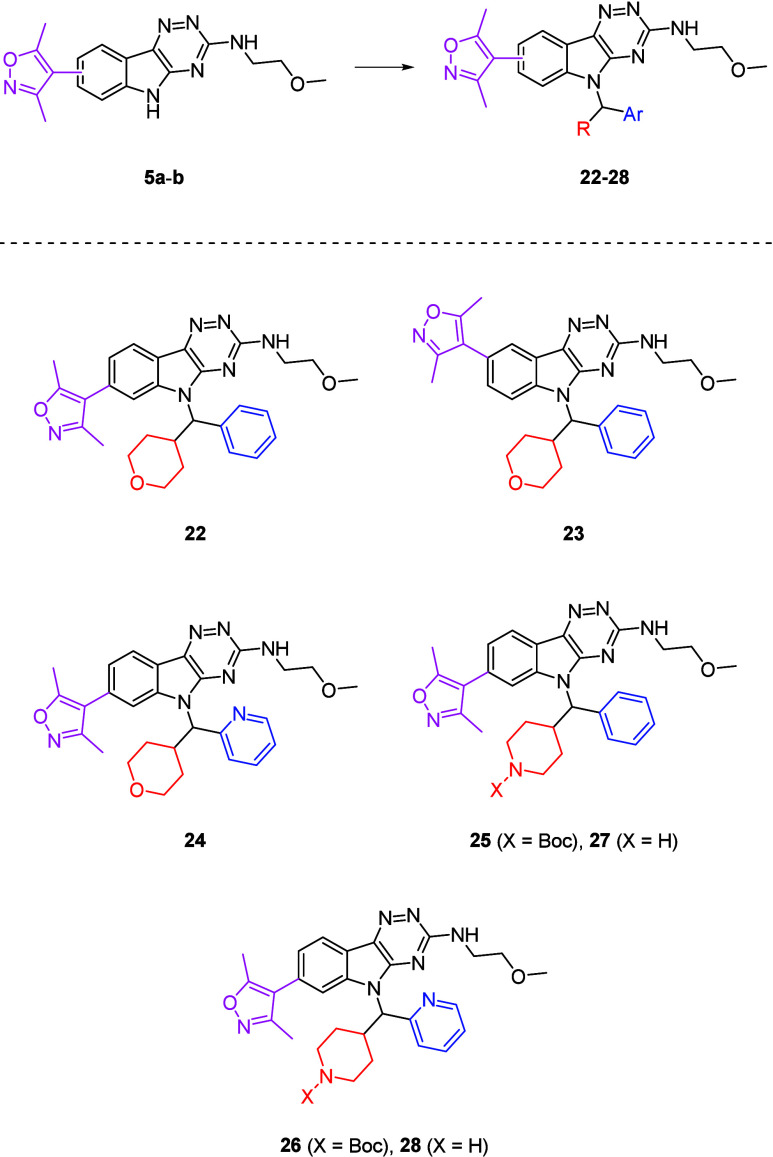 Figure 11
Figure 11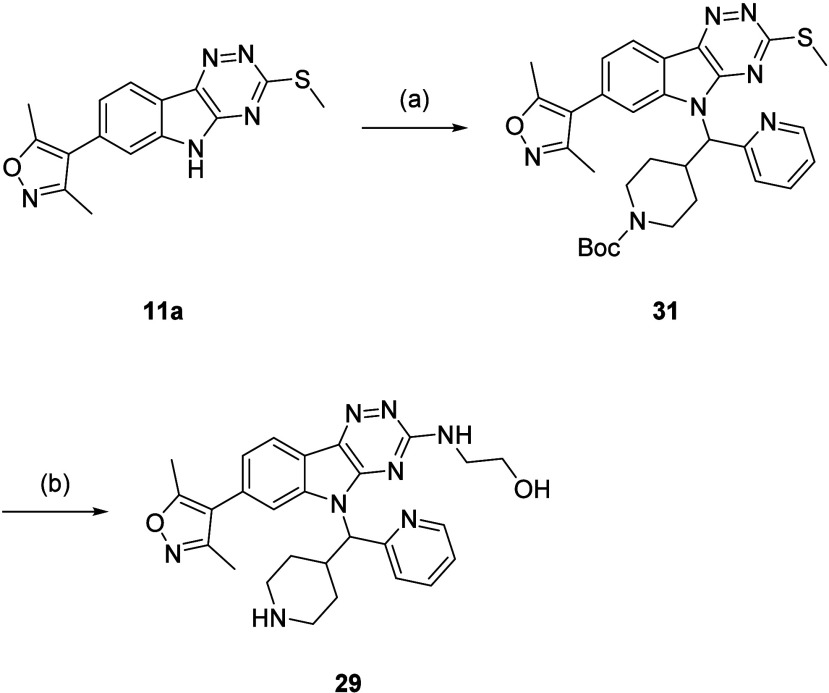 Figure 12
Figure 12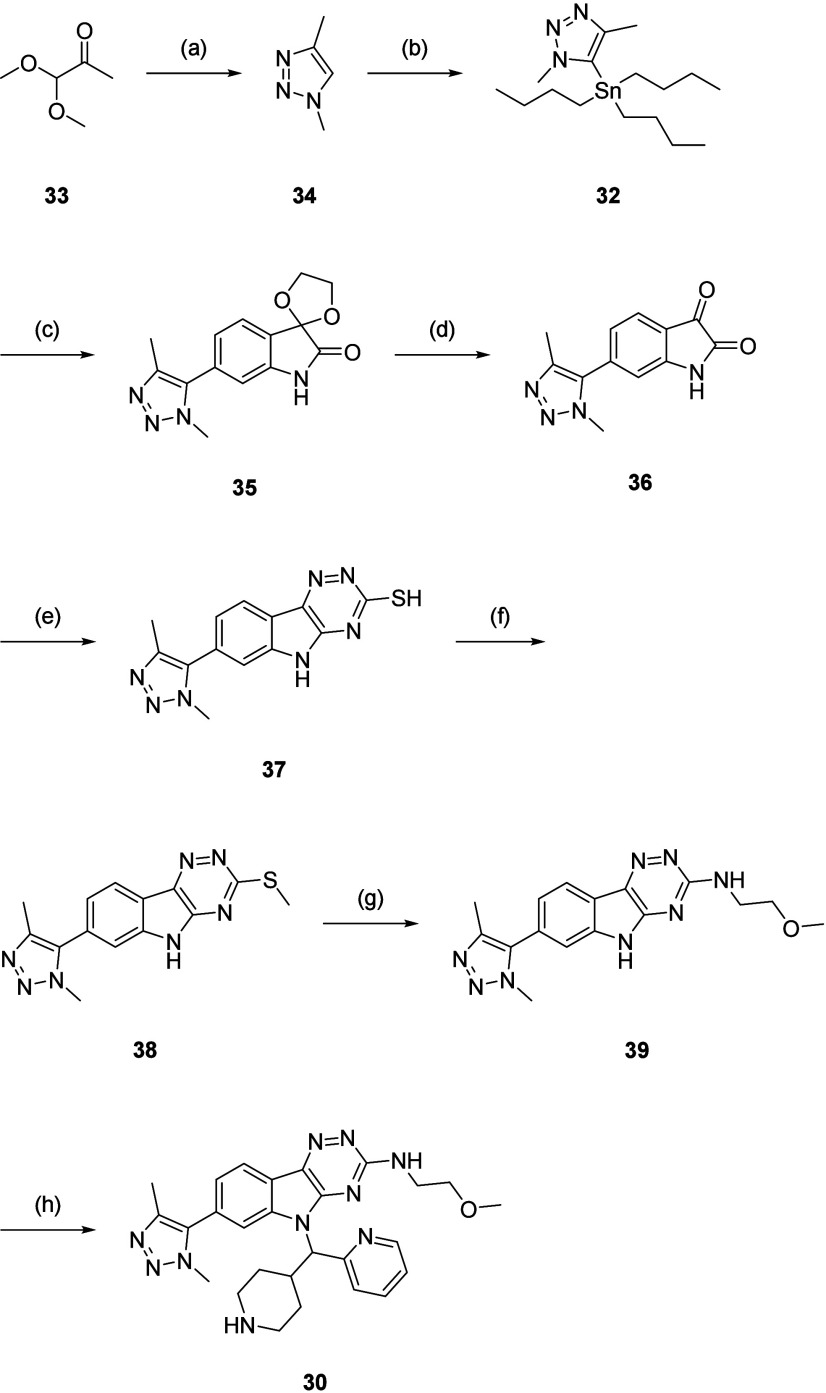 Figure 13
Figure 13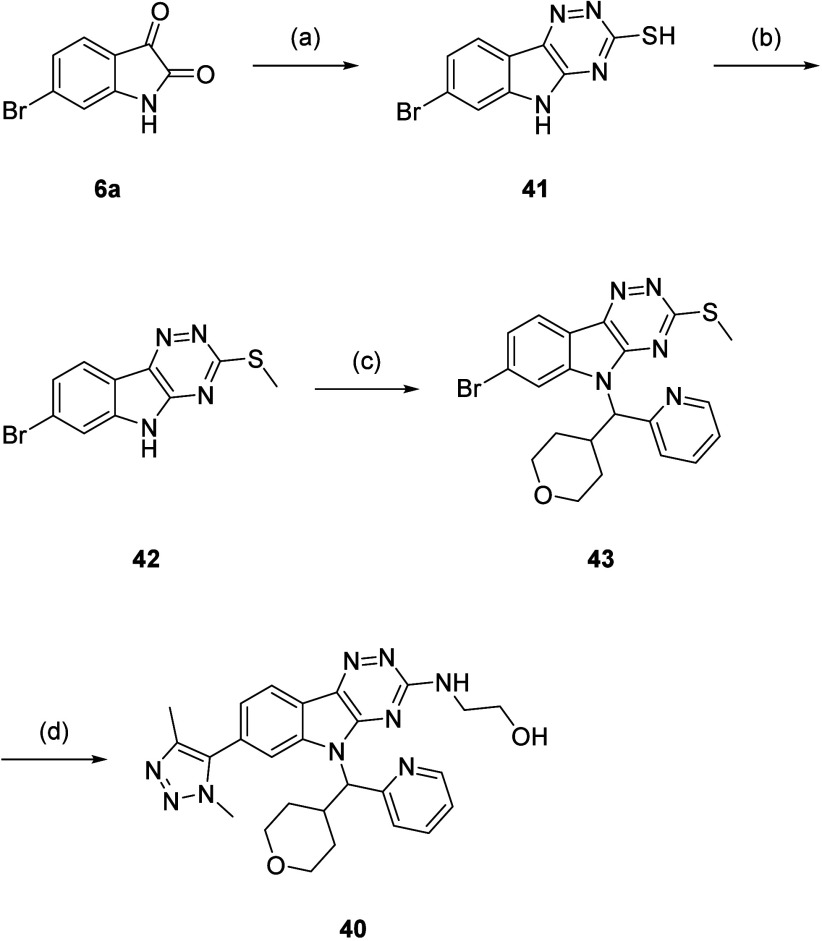 Figure 14
Figure 14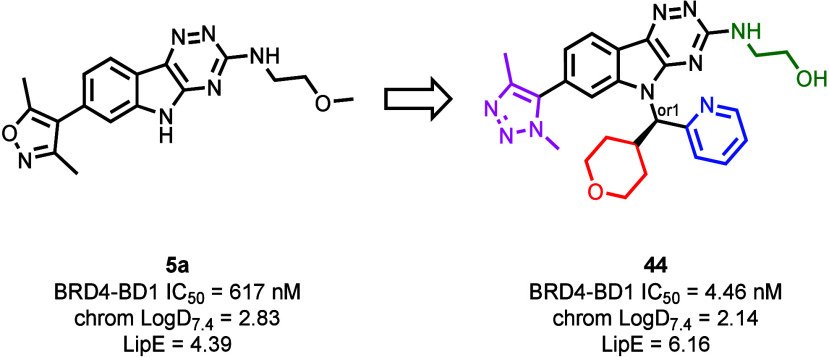 Figure 15
Figure 15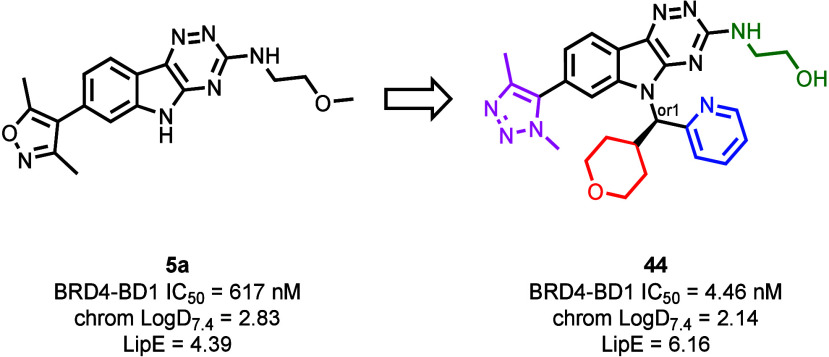 Figure 16
Figure 16- —GlaxoSmithKline10.13039/100004330
- —Engineering and Physical Sciences Research Council10.13039/501100000266
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsProtein Degradation and Inhibitors · Multiple Myeloma Research and Treatments · Ubiquitin and proteasome pathways
The bromodomain and extra-terminal domain (BET) protein family is composed of bromodomain-containing proteins 2, 3, and 4 (BRD2, BRD3 and BRD4 respectively) and bromodomain testis-specific protein (BRDT).? These proteins are epigenetic readers that form multimolecular complexes with enzymes involved in the covalent modification of DNA and the proteins that package DNA, such as histones, and so play an important role in several physiological processes.?
The primary function of BETs is to regulate cell transcription by binding to N-acetylated lysine residues on histone tails.? BET inhibitors prevent this key interaction in cancer cells, triggering the downregulation of certain genes and resulting in an overall reduction in gene expression.? There has been extensive research in recent years, resulting in the successful development of over 20 small molecule BET inhibitors, with some of these now in clinical trials for the treatment of hematological malignancies and solid tumors.? While preliminary results have confirmed the antitumor potential of BET inhibitors, several programs have also been terminated due to either safety concerns or limited monotherapeutic efficacy.?
There are several bioisosteres which can be incorporated into BET inhibitor scaffolds to displace N-acetylated lysine-mimicking peptides from bromodomains (BDs) (Figure).? The first BET inhibitor was JQ1 (1), reported in 2010.? This compound includes a methyltriazole moiety, which acts as an N-acetylated lysine mimetic. Other representative examples are the dimethylisoxazole-containing I-BET151 (2) and the pyridone-based ABBV-075 (3). ?,? Isoxazoles are of particular interest as N-acetylated lysine mimetics due to their structural simplicity and high lipophilic efficiency (LipE).? However, it can be challenging to develop isoxazole-containing BET inhibitors with high potency, as seen in the case of 2.?
Compounds 1, 2 and 3 effectively downregulate cellular myelocytomatosis (c-Myc) expression and inhibit tumor cell proliferation when used to treat advanced malignancies such as AML. ?−? ? Unfortunately, 1 had low oral bioavailability and overly short half-life of 1 h and was therefore unable to progress to clinical trials.? 2 exhibited cardiotoxicity in Phase I studies and 3 is currently undergoing evaluation, underlining the difficulties in advancing BET inhibitors through clinical trials. ?−? ?
The final BET inhibitor discussed is BMS-986158 (4) (Figure).? 4 has high potency (pIC_50_ = 8.54) against BRD4-BD1, with a dimethyltriazole ring as the N-acetylated lysine mimetic. However, due to high lipophilicity (cLogP ∼ 5.0), it has a low LipE of 3.51. ?,? LipE is an important parameter used in drug discovery linking potency and lipophilicity and is calculated from pIC_50_ minus cLogP.? Empirical evidence suggests that drug candidates have a LipE > 6, obtained by increasing potency while decreasing lipophilicity during lead optimization giving compounds with better pharmacological properties, such as improved solubility and metabolic stability.?
Our aim was to develop a BET inhibitor with levels of potency comparable to those of 4 with reduced lipophilicity (increased LipE). A new tricyclic scaffold was prepared, and we report its optimization to highly potent and metabolically stable BET inhibitors.
We began with the identification of a triazinoindole core, inspiration of which originated from previous research carried out by Soukarieh which focused on developing novel treatments for Pseudomonas aeruginosa.? The dimethylisoxazole moiety was incorporated as an N-acetylated lysine mimetic, located at either the 7- (5a) or 8- (5b) positions (Figure).? The 2-methoxyethylamine group on the triazine was included to aid in aqueous solubility. Docking analysis of the BRD4-BD1 binding pocket confirmed that the WPF shelf, an important tryptophan-proline-phenylalanine sequence found in all BRDs, could be accessed from functionalization of the indole nitrogen given the structural similarity of 5 to benchmark 4 (Figures S1–2). ?,?
The synthesis of 5a-b commenced with acetal protection of either 6-bromoisatin (6a) or 5-bromoisatin (6b), followed by Suzuki–Miyaura cross-coupling with 3,5-dimethylisoxazole-4-boronic acid pinacol ester (Bpin) to afford biaryls (8a-b) (Scheme). Acid-mediated deprotection gave isatins (9a-b). Cyclization of these with thiosemicarbazide formed triazines (10a-b) and S-methylation yielded thioethers (11a-b). A one-pot oxidation and displacement process provided 5a-b. N-Alkylations using the requisite alkyl halide gave N-alkylindoles (12-13) (Scheme).
5a-b and 12-13 were submitted for biological evaluation with three main metrics measured, potency (pIC_50_), LipE and property forecast index (PFI) (Table). Potency was determined against BRD4-BD1 using FRET binding, with this being treated as a surrogate of BRD4 activity given the homology between BD1 and BD2 across the BET proteins. ?,? PFI is calculated from hydrophobicity plus aromatic ring count (chromatographic (chrom) logD_7.4_ + #Ar), with this metric being indicative of developability, or likelihood of progression to a drug candidate.? PFI was used to assess the physicochemical profile of compounds, with a target of PFI < 7 as risks for several parameters, including solubility, are increased above this upper limit.? Our objective was to develop a BET inhibitor with high potency and favorable physicochemical properties, as determined using PFI and LipE to guide compound optimization.
Initial results showed the 7-substituted analogues displayed higher biological activities than their 8-substituted counterparts, with this difference amplified upon further functionalization. 5a was approximately four times more potent than its matched pair 5b (pIC_50_ = 6.22 for 5a, versus 5.64 for 5b). Compounds 12-13 all demonstrated submicromolar activity against BRD4-BD1, and this observation was consistent with literature precedent that a lipophilic functional group in the vicinity of the WPF shelf is optimal for binding to the BRD.? Unfortunately, the high lipophilicity of these molecules meant that they had PFI > 7 and so were likely to have solubility issues.?
We focused on reducing PFI and increasing LipE to more acceptable levels, and this was achieved by attaching a branched substituent containing an aromatic group to boost potency, through interaction with the WPF shelf, and an aliphatic group to lower lipophilicity, measured as chrom LogD_7.4_. The method of N-functionalization employed was a Mitsunobu reaction, and preparation of the corresponding alcohols was achieved in two steps. The unsymmetrical ketone precursors (14-17), obtained from a reaction between the constituent aliphatic ketones and aryl aldehydes, were reduced to the alcohols (18-21) (Scheme).? Subsequent Mitsunobu reactions gave N-alkylindoles (22-26) (Scheme). Given the differences in experimental potency, priority was given to synthesizing 7-substituted analogues, derived from 5a. In the cases of 25 and 26, the Boc protecting groups were removed to give N-alkylindoles (27-28).
Compounds 22-24 and 27-28 were biologically evaluated (Table). Some compounds proved potent (IC_50_ < 10 nM), an encouraging feat given the previously outlined difficulties in developing isoxazole-containing BET inhibitors with potencies above the micromolar range.? 28 represented the most favorable balance of potency, PFI and LipE (pIC_50_ = 7.74, PFI = 7.37 and LipE = 4.25). The incorporation of a piperidine moiety proved beneficial as a chrom LogD_7.4_ modulator with the PFI closer to 7. Effort was now focused on reducing PFI < 7 and increasing LipE in 28.
Two structural modifications to 28 proposed to further lower lipophilicity were to replace the methoxy ethylamine with a hydroxy ethylamine and change the lipophilic dimethylisoxazole for a more polar dimethyltriazole. This led to the design of N-alkylindoles (29-30) (Figure). The isoxazole-triazole switch was not expected to impact potency given the similarity in binding modes between the two N-acetylated lysine mimetics with BRD4-BD1, with the same strategy adopted by Gavai during the development of 4.?
Demethylation of 28 to produce 29 was not possible due to difficulties in carrying out these reactions on aliphatic methyl ethers.? Instead, the synthetic route that was previously used in the preparation of 5a-b was used here to make 11a, at which point a Mitsunobu reaction with 21 was carried out to attain N-alkylindole (31) (Scheme). This was subjected to one-pot oxidation and displacement with ethanolamine, followed by Boc deprotection, and gave 29.
For 30, dimethyltriazole (32) was required. A one-pot, three-component coupling of 1,1-dimethoxyacetone (33), TsNHNH_2_ and MeNH_2_ yielded dimethyltriazole (34), which was converted to 32 through lithiation and reaction with Bu_3_SnCl (Scheme).? Stille coupling of 7a with 32 afforded 35 and acid-mediated acetal deprotection yielded isatin (36). Cyclization with thiosemicarbazide formed triazine (37), which was S-methylated to give thioether (38). A one-pot oxidation and displacement with 2-methoxyethylamine produced an amine (39). A Mitsunobu reaction of 39 with 21, followed by the final Boc deprotection, gave 30.
The biological data for 29 and 30 was encouraging, with both demonstrating high potencies against BRD4-BD1 (pIC_50_ = 7.29 ± 0.06 for 29 (LipE = 4.23) and 7.41 ± 0.03 for 30 (LipE = 4.93)). The PFI was also <7 (PFI = 6.74 for 29 and 6.46 for 30), indicating that the structural modifications to the core scaffold had been beneficial. Additionally, the in vitro permeability of these compounds was determined by a parallel artificial membrane permeability assay (PAMPA).? Unfortunately, both molecules possessed very low permeabilities (<3.00 nm/s), which was hypothesized to be due to the basic piperidine moiety. This would be protonated at physiological pH and decrease the overall affinity of the compound toward the hydrophobic membrane region.?
The emphasis switched to improving permeability, and this was achieved by combining the moieties which were expected to give the best balance of potency and physicochemical properties. The hydroxy ethylamine side chain in 29 and triazole warhead in 30 were incorporated, and the piperidine moiety was replaced with a polar nonbasic THP ring to give N-alkylindole (40) (Figure). Given the permeability of 4 (498 nm/s), which has a similar core, and the potency of our closest matched pair 24 (pIC_50_ = 8.01), which contains the same WPF shelf group, we were sufficiently encouraged that these structural changes would increase permeability without compromising potency.
The synthesis of 40 started from 6a, with cyclization using thiosemicarbazide forming triazine (41) (Scheme). S-Methylation gave thioether (42), and a Mitsunobu reaction yielded N-alkylindole (43). One-pot oxidation and displacement with ethanolamine were followed by Stille coupling using 32 to afford 40.
Compound 40 showed increased potency and LipE compared to 29 and 30 (pIC_50_ = 7.98 ± 0.17 (LipE = 5.79) for 40, compared to 7.29 (LipE = 4.23) and 7.41 (LipE = 4.93) for 29 and 30, respectively). As expected, there was a slight increase in PFI due to the piperidine-THP switch (PFI = 7.15 for 40, compared to 6.74 and 6.46 for 29 and 30, respectively). The permeability of 40 was significantly better (104 nm/s, compared to <3.00 nm/s for both 29 and 30), due to the reduced hydrogen bond donor count and absence of ionizable center in 40 compared to 29 and 30. These results provided validation that racemate 40 was the lead compound as it had the best balance of potency and physicochemical properties, and this was purified by chiral HPLC (CHIRALPAK IB N-5 column) to yield enantiomers (44) and (45) (Figure).
Compound 44 (pIC_50_ = 8.35 ± 0.01) was more active compared to 45 (pIC_50_ = 8.02 ± 0.08), while the PFIs of both enantiomers were identical (PFI = 7.14). For both enantiomers, kinetic solubility, permeability and metabolic stability data, the latter measured by in vitro microsomal intrinsic clearance (CL_int_) in rats and humans, was collected (Table). ?,?,? Docking of 44 and 45 into the BRD4-BD1 binding pocket was also carried out (Figures S3–4).
The enantiomers had comparable solubilities (134 μg/mL for 44 and 138 μg/mL for 45), and permeabilities (77.8 nm/s for 44 and 93.6 nm/s for 45). With CL_int_ < 10.0 μL/min/mg of protein in rats and humans, both compounds could be considered metabolically stable.? Our lead compound, 44 showed levels of activity analogous to those of 4 (pIC_50_ = 8.54), while PFI was reduced (PFI = 7.14 for 44, compared to 9.08 for 4). As both compounds contain 5 aromatic rings, this difference in PFI resulted from an advantageous decrease in chrom LogD_7.4_ (2.14 for 44, compared to 4.08 for 4), as high lipophilicity has been shown to correlate with attrition through the drug development phases.?
Our SAR optimization strategy throughout this work consisted of improving potency and reducing lipophilicity. Plotting pIC_50_ against cLogP for a range of compounds allows a series to be ranked to suggest molecules with improved physicochemical properties. This was carried out for all 16 analogues, with 4 included as a comparator (Figure). Compound 44 (LipE = 6.16) demonstrated a significantly higher LipE than 4 (LipE = 3.51).
In conclusion, a novel triazinoindole scaffold for targeted BET inhibition was synthesized using a traditional SAR approach. Late-stage functionalization gave a series of N-substituted indole derivatives. Preliminary results indicated that many had submicromolar levels of potency but retained the characteristically high PFI of several documented BET inhibitors. The second iteration, implemented primarily to reduce PFI and improve LipE by making strategic structural modifications to the core scaffold, afforded 29 and 30, both of which had PFIs < 7. However, their progression was hindered by poor membrane permeabilities. Further optimization yielded 40, which demonstrated nanomolar potency against BRD4-BD1 and had an acceptable PFI of 7.15. 40 was separated to produce enantiomers 44 and 45, with 44 being more potent. 44 displayed comparable activity against BRD4-BD1 to clinical candidate 4 and possessed lower lipophilicity, as demonstrated by an improvement in LipE. The structurally close homologue (compound 4) was reported to display pan-inhibitory effects on binding to BRD2, BRD3, and BRD4 (IC_50_ = 0.8, 1.4, and 1.1 nM, respectively) along with potent cytotoxicity, consistent with the role of c-Myc in driving proliferation in a panel of human lung cancer cell lines.? Further work is ongoing in our laboratories to confirm BET-dependent cellular effects for 44, and these results will be communicated in due course.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Morgado-Pascual J. L.Rayego-Mateos S.Tejedor L.Suarez-Alvarez B.Ruiz-Ortega M.Bromodomain and Extraterminal Proteins as Novel Epigenetic Targets for Renal Diseases Front. Pharmacol.201910131510.3389/fphar.2019.0131531780938 PMC 6857099 · doi ↗ · pubmed ↗
- 2Arrowsmith C. H.Bountra C.Fish P. V.Lee K.Schapira M.Epigenetic protein families: a new frontier for drug discovery Nat. Rev. Drug Discovery 201211538440010.1038/nrd 367422498752 · doi ↗ · pubmed ↗
- 3Zhou Z.Li X.Liu Z.Huang L.Yao Y.Li L.Chen J.Zhang R.Zhou J.Wang L.Zhang Q.-Q.A Bromodomain-Containing Protein 4 (BRD 4) Inhibitor Suppresses Angiogenesis by Regulating AP-1 Expression Front. Pharmacol.202011104310.3389/fphar.2020.0104332765266 PMC 7381267 · doi ↗ · pubmed ↗
- 4Delmore J. E.Issa G. C.Lemieux M. E.Rahl P. B.Shi J.Jacobs H. M.Kastritis E.Gilpatrick T.Paranal R. M.Qi J.Chesi M.Schinzel A. C.Mc Keown M. R.Heffernan T. P.Vakoc C. R.Bergsagel P. L.Ghobrial I. M.Richardson P. G.Young R. A.Hahn W. C.Anderson K. C.Kung A. L.Bradner J. E.Mitsiades C. S.BET bromodomain inhibition as a therapeutic strategy to target c-Myc Cell 2011146690491710.1016/j.cell.2011.08.01721889194 PMC 3187920 · doi ↗ · pubmed ↗
- 5Lu T.Lu W.Luo C.A patent review of BRD 4 inhibitors (2013–2019)Expert Opin. Ther. Pat.2020301578110.1080/13543776.2020.170264531815566 · doi ↗ · pubmed ↗
- 6Wang Z.-Q.Zhang Z.-C.Wu Y.-Y.Pi Y.-N.Lou S.-H.Liu T.-B.Lou G.Yang C.Bromodomain and extraterminal (BET) proteins: biological functions, diseases and targeted therapy Signal Transduct. Target. Ther.20238142010.1038/s 41392-023-01647-637926722 PMC 10625992 · doi ↗ · pubmed ↗
- 7Hewings D. S.Wang M.Philpott M.Fedorov O.Uttarkar S.Filippakopoulos P.Picaud S.Vuppusetty C.Marsden B.Knapp S.Conway S. J.Heightman T. D.3,5-Dimethylisoxazoles Act As Acetyl-lysine-mimetic Bromodomain Ligands J. Med. Chem.201154196761677010.1021/jm 200640 v 21851057 PMC 3188285 · doi ↗ · pubmed ↗
- 8Filippakopoulos P.Qi J.Picaud S.Shen Y.Smith W. B.Fedorov O.Morse E. M.Keates T.Hickman T. T.Felletar I.Philpott M.Munro S.Mc Keown M. R.Wang Y.Christie A. L.West N.Cameron M. J.Schwartz B.Heightman T. D.La Thangue N.French C. A.Wiest O.Kung A. L.Knapp S.Bradner J. E.Selective inhibition of BET bromodomains Nature 201046873271067107310.1038/nature 0950420871596 PMC 3010259 · doi ↗ · pubmed ↗