Dataset on effect of cadmium and lead on chemical reactivity of selected heterocyclic compounds as potential dipeptidyl peptidase III inhibitors using insilico method
Kehinde Adeola Bolaji, Abel Kolawole Oyebamiji, Godwin Oladele Olutona

TL;DR
This paper investigates how cadmium and lead affect the chemical reactivity of heterocyclic compounds that could inhibit an enzyme called dipeptidyl peptidase III.
Contribution
The study introduces a computational analysis of metal effects on heterocyclic compounds' reactivity and drug potential.
Findings
Cadmium significantly influenced the HOMO energy and energy gap of compound 7.
Cadmium altered the LUMO energy of compound 5.
Pharmacokinetic properties of high-affinity ligands were compared to reference molecules.
Abstract
The role played by heterocyclic compounds in drug design and discovery remain crucial globally. In this work, the effect of cadmium and lead were investigated on the chemical reactivity of selected heterocyclic compounds with potential anti-dipeptidyl peptidase III activity using insilico method. Various software such as Spartan 14 (for optimization), molecular operating environment (moe) (for induced fit docking) and ADMETSar (pharmacokinetic examination) were used to execute the investigations. Three descriptors (highest occupied molecular orbital energy (EHOMO); lowest unoccupied molecular orbital energy (ELUMO) and energy gap) were observed among others for selected compounds without any foreign attachment, selected compounds with cadmium and selected compounds with lead. The calculated descriptors revealed that cadmium had a great effect on compound 7 in term of HOMO energy and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
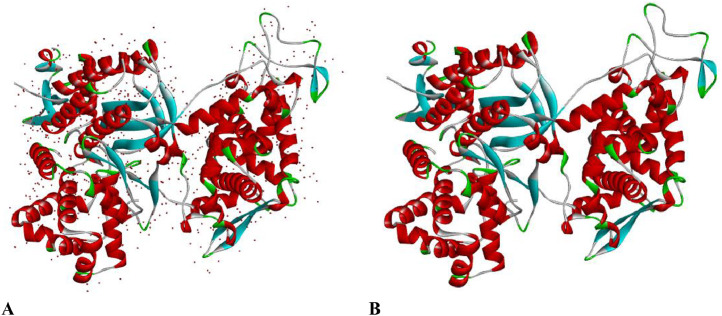 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsComputational Drug Discovery Methods · Neuropeptides and Animal Physiology · Peptidase Inhibition and Analysis
Specifications TableSubjectInsilico StudySpecific subject areaDrug EvaluationType of dataFigure, Chart, Table, ADMET, GraphData collectionThe investigated compounds (compound without foreign atom; compound + cadmium and compound + lead) were theoretically calculated using Spartan 14 in order to have access to the features encoded in the examined compounds. The compounds with no foreign atom attached were docked into the active site of Dipeptidyl Peptidase III [pdb id: 3fvy] using induced fit method via molecular operating environment software. The role of retrieved descriptors from theoretically calculated compounds in the inhibiting ability of the examined compounds were investigated using Microsoft excel software and pharmacokinetics evaluation was exhibited on compound with highest binding affinity. Every outcome was reported and described.Data source locationComputational Chemistry Research Laboratory, Department of Chemistry and Industrial Chemistry, BOWEN University, PMB 284, Iwo, Osun State, Nigeria.Data accessibilityhttps://data.mendeley.com/datasets/wrhd27phzc/1Repository name: Mendeley DataData identification number: 10.17632/wrhd27phzc.1Direct URL to data: https://data.mendeley.com/datasets/wrhd27phzc/1Instructions for accessing these data: The data can be accessed using the above URLRelated research articleNone
Value of the Data
1
- •The calculated descriptors will show to scientists the effect of cadmium and lead on the geometries of the compounds under study.
- •The two dimensional structure of the examined compounds will expose to researchers the nature of the bond between atoms used in the work.
- •The configuration of the raw and treated receptors will be exposed to researchers.
- •The calculated features for individual compound will reveal to researchers the type of reaction the examined compound can be involved in.
- •The scoring, the calculated distance, the predicted bond type and amino acid residue will show to scientists the nature of interactions involved in the examined complexes.
- •The results of the pharmacokinetic analysis will provide information on the absorption, digestion, metabolism, and excretion processes of the molecule with the highest binding affinity in the human system.
Background
2
The objectives of this work are:
- ➢To investigate the biological effects of cadmium and lead on geometries (bond length and bond angle) of the optimized selected heterocyclic compounds.
- ➢To evaluate the biochemical efficiency of the optimized selected heterocyclic compounds docked against dipeptidyl peptidase III [pdb id: 3fvy] in order to down-regulate pain in mammals.
- ➢To identify the ability of the compound with highest binding affinity to act as drug-agent via pharmacokinetics evaluation.
Data Description
3
The 2-dimensional and 3-dimenional structure of the examined compound as well as the IUPAC name of the examined compound were presented on Table 1. As shown in Table 1, the 3-dimensional structure of the examined compounds comprises of atom which were represented by using different ball (red=oxygen; blue=Nitrogen; yellow = Sulphur) were bonded together to form the compounds under investigation.Table 12-Dimensional and 3-dimensional Structures and IUPAC name of the Investigated compounds.Table 1:2-D StructureIUPAC Names1Image, table 12-acetoxybenzoic acid2Image, table 13-ethyl 5-methyl 2-((2-aminoethoxy)methyl)-4-(2-chlorophenyl)-6-methyl-1,4-dihydropyridine-3,5-dicarboxylate3Image, table 1N4-(7-chloroquinolin-4-yl)-N1,N1-diethylpentane-1,4-diamine4Image, table 13-(4-chlorophenyl)-N,N-dimethyl-3-(pyridin-2-yl)propan-1-amine5Image, table 11-cyclopropyl-6-fluoro-4-oxo-7-(piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid6Image, table 11-((2-chlorophenyl)diphenylmethyl)-1H-imidazole7Image, table 12-(2-methyl-5-nitro-1H-imidazol-1-yl)ethan-1-ol8Image, table 1nicotinamide9Image, table 1N-(4-hydroxyphenyl)acetamide10Image, table 14-hydroxy-2-methyl-N-(pyridin-2-yl)-2H-benzo[e][1,2]thiazine-3-carboxamide 1,1-dioxide
Table 2 showed the calculated features (highest occupied molecular orbital energy (EHOMO), lowest unoccupied molecular orbital energy (ELUMO) and energy gap) for the optimized compound, optimized compound + cadmium and optimized compound + lead. The graphical representation of the HOMO and LUMO orbital energy profile for the entire compound investigated in this work was displayed in Table 3.Table 2. Selected features for the optimized compounds.Table 2:Selected CompoundSelected Compound + CadmiumSelected Compound + LeadHOMO (eV)LUMO (eV)Energy Gap (eV)HOMO (eV)LUMO (eV)Energy Gap (eV)HOMO (eV)LUMO (eV)Energy Gap (eV)1-10.27-1.209.07-7.23-2.534.70-10.28-1.229.062-8.75-0.478.28-7.56-2.764.80-8.78-0.498.293-8.55-0.957.60-7.39-2.594.80-8.55-0.957.604-8.35-0.228.13-7.48-2.704.78-8.36-0.248.125-9.01-0.998.02-6.41-4.571.84-9.01-0.998.026-9.22-0.498.73-7.70-2.894.81-9.23-0.498.747-10.65-2.048.61-6.03-4.261.77-10.63-2.068.578-10.52-0.899.63-7.68-2.894.79-10.51-0.889.639-9.17-0.069.11-7.83-3.024.81-9.17-0.059.1210-9.43-1.497.94-7.83-3.024.81-9.43-1.507.93Table 3Predicted orbital energy profile for the selected compounds.Table 3:Selected CompoundSelected Compound + CadmiumSelected Compound + Lead1Image, table 3Image, table 3Image, table 32Image, table 3Image, table 3Image, table 33Image, table 3Image, table 3Image, table 34Image, table 3Image, table 3Image, table 35Image, table 3Image, table 3Image, table 36Image, table 3Image, table 3Image, table 37Image, table 3Image, table 3Image, table 38Image, table 3Image, table 3Image, table 39Image, table 3Image, table 3Image, table 310Image, table 3Image, table 3Image, table 3
Fig. 1 revealed raw Dipeptidyl Peptidase III and treated Dipeptidyl Peptidase III. Fig. 1A showed the investigated receptor with water molecules and other foreign small molecules while Fig. 1B showed the cleaned Dipeptidyl Peptidase III. Also, Table 4 showed the predicted binding sites obtained from the treated Dipeptidyl Peptidase III. The factors considered were size, protein ligand-binding (PLB), Hyd, side and residues.Fig. 13-dimensional structure of (A) raw Dipeptidyl Peptidase III (B) treated Dipeptidyl Peptidase III.Fig 1:Table 4. The calculated binding site for dipeptidyl peptidase III [pdb id: 3fvy].Table 4:SiteSizePLBHydSideResidues15444.761342641:(GLN5 ILE7 LEU8 PRO9 ASP11 ILE12 GLY13 ASP18 CYS19 ARG20 GLU21 ALA22 TRP42 VAL48 TYR100 SER101 ASN102 MET103 SER108 PHE109 GLY110 ASP111 TYR171 GLU316 SER317 TYR318 ARG319 GLU327 PHE328 GLU329 VAL334 VAL335 ASN336 MET339 SER340 LYS342 PHE343 ASP372 PHE373 THR374 SER375 LEU376 ASP377 PHE381 ALA382 GLY383 SER384 GLY385 ILE386 PRO387 ALA388 ASN406 VAL4 ... AL673 GLN674 LEU696 SER699 PHE700 ARG703)2470.1318451:(TYR35 SER38 ARG39 TRP42 THR380 PRO675 ASN676 THR677 ARG678 LEU679 GLU702 ARG703 PHE704 PRO705 GLU706)3850.0727541:(TYR58 ALA61 ARG65 ARG68 ALA69 ALA132 GLN135 HIS136 GLU139 VAL140 GLY142 LEU143 THR146 PRO709 GLU712 GLU713 THR716)4500.0519331:(TYR60 LEU63 SER64 PHE67 ARG68 TYR100 ILE697 PHE700 SER701 GLU702 ARG703 PHE704 PRO705 GLU706 ASP707 GLY708 PRO709 GLU712)537-0.114191:(ARG421 GLU422 LYS423 LEU424 THR425 PHE426 LEU427 GLU429 LYS432 ASP600 ALA601 ARG602 ARG665)635-0.1815291:(ASN192 LEU193 SER194 ASN197 LEU216 GLU229 VAL230 LYS233 GLY288 SER289 ILE290 GLU291 LYS294)711-0.197311:(TYR35 HIS36 ARG39 TYR43 LYS257 GLU260 GLN261 LYS264 LEU679 GLU706 ASP707)810-0.215151:(ARG215 ARG248 GLY249 ASP250 TYR251 LEU718 ALA721 ASP722)945-0.2119291:(ASP303 LYS304 GLY305 PRO306 GLU309 VAL335 ASP372 THR374 LEU376 PHE404 LYS405 ASN406 VAL407)1039-0.2210221:(THR501 ILE502 SER505 TYR557 ALA561 PHE562 ASN563 TRP564 GLN570 GLU642 ALA645 THR646)1123-0.229211:(SER194 TYR196 ILE290 LYS294 ASN394 TYR395 ASP396 ASP397)1231-0.2213211:(GLU351 LEU354 LYS355 GLU356 LEU357 PRO358 TRP359 PRO360 PRO361 GLU364 LYS365 ASP366)1321-0.237181:(ASN10 ASP11 ILE12 VAL14 GLN90 VAL94 GLN420 GLU422 LYS423 ALA691 SER692 ALA693)1411-0.2513231:(ARG577 LEU580 GLU581 TYR644 GLU653 PHE655)1541-0.2614211:(GLY305 VAL335 ASN336 MET339 SER340 PHE343 PHE373 THR374 SER375 LEU376)1615-0.2613211:(LEU49 LEU50 SER53 PRO54 PRO57 THR170 ARG199 LEU718 ALA719 ASP722 TRP726)1715-0.278151:(VAL578 GLU581 ALA582 ALA617 ARG620 PHE621 TYR644)1819-0.2810211:(TYR196 ILE315 GLU316 SER317 TYR318 ASN394)1910-0.294141:(GLU428 ASP430 ASP431 TYR540 LEU544 VAL603 ARG604 LEU605 ASP606)2016-0.3011171:(LYS365 ASP366 LYS367 LEU369 LYS459)2123-0.3112231:(GLU316 TYR318 GLU329 PHE381 PRO387 GLY389 ILE390 ASN391 ILE392 ASN394)2216-0.338171:(GLN5 LYS107 PHE109 THR112 SER668 ARG669)236-0.33381:(TYR196 SER317 GLY323 SER324)246-0.333111:(GLU713 THR716 GLN717 THR720)
The calculated binding affinity for the examined heterocyclic compounds and Dipeptidyl Peptidase III [pdb id: 3fvy] were -3.92 kcal/mol for compound 1, -6.39kcal/mol for compound 2, -6.43 kcal/mol for compound 3, -6.25 kcal/mol for compound 4, -6.42kcal/mol for compound 5, -5.57 kcal/mol for compound 6, -3.70 kcal/mol for compound 7, -7.08 kcal/mol for compound 10 and -5.29 kcal/mol for reference compound. As displayed in Table 5, no result were generated for compound 8- Dipeptidyl Peptidase III [pdb id: 3fvy] and compound 9- Dipeptidyl Peptidase III [pdb id: 3fvy] complexes.Table 5. Calculated scoring for compound 1-10 in kcal/mol.Table 5:Scoring (kcal/mol)1-3.922-6.393-6.434-6.255-6.426-5.577-3.708-9-10-7.08Ref-5.29Ref: Ibuprofen
More so, to access the efficiency of the ligand to inhibit Dipeptidyl Peptidase III [pdb id: 3fvy], distance, bond type, residue, 2-dimensional configuration of the docked compound in the active site of the receptor were considered (Table 6). The calculated nonbonding interaction were 3.65, 2.65, 3.19 and 2.65 (Distance); H-acceptor, H-acceptor, Ionic and Ionic (bond type) and GLN 261, LYS 264, ARG 39 and LYS 264 (Residue) for compound 1; 2.85, 2.85 and 3.23 (Distance); H-donor, Ionic and Ionic (bond type); GLU 344, GLU 344 and GLU 344 (Residue) for compound 2; 3.29, 3.29, 3.95, and 3.97 (Distance); H-donor, Ionic, Ionic and Ionic (bond type); GLU 329, GLU 329, ASP 111, ASP 111 (Residue) for compound 3; 3.16, 3.16 and 4.52 (Distance); H-donor, Ionic and pi-H (bond type); GLU 327, GLU 327 and PRO 387 (Residue) for compound 4; 3.28, 3.26, 3.04, 2.86, 3.04, 2.86, 3.85, 3.28 and 4.19 (Distance); H-donor, H-acceptor, H-acceptor, H-acceptor, Ionic, Ionic, Ionic, Ionic and H-pi (bond type); GLU 451, LYS 629, LYS 629, LYS 365, LYS 629, LYS 365, LYS 629, GLU 451and HIS 455 (Residue) for compound 5; 3.05, 3.15 and 3.68 (Distance); H-acceptor, H-acceptor and pi-H (bond type); ASN 10, ASN 117 and PRO 9 (Residue) for compound 7; 2.76 and 3.11 (Distance); H-acceptor and H-acceptor (bond type); SER 375 and ASN 336 (Residue) for compound **10.**Table 6. Selected non-bonding interaction and visual representation for the investigated complexes.Table 6:LigandDistanceBond TypeResidue2D Diagram13.65H-acceptorGLN 261Image, table 62.65H-acceptorLYS 2643.19IonicARG 392.65IonicLYS 26422.85H-donorGLU 344 (A)Image, table 62.85IonicGLU 344 (A)3.23IonicGLU 344 (A)33.29H-donorGLU 329 GLU 329Image, table 63.29IonicASP 1113.95IonicASP 1113.97Ionic43.16H-donorGLU 327Image, table 63.16IonicGLU 3274.52pi-HPRO 38753.28H-donorGLU 451Image, table 63.26H-acceptorLYS 6293.04H-acceptorLYS 6292.86H-acceptorLYS 3653.04IonicLYS 6292.86IonicLYS 3653.85IonicLYS 6293.28IonicGLU 4514.19H-piHIS 4556---Image, table 673.05InteractionASN 10Image, table 63.15H-acceptorASN 1173.68H-acceptorPRO 9pi-H8----9----102.76InteractionSER 375Image, table 63.11H-acceptorASN 336H-acceptor
Table 7, Table 8 showed the pharmacokinetic analysis of the lead compound (compound 10) and Ibuprofen (reference compound). The analysis was presented in ADMET predicted profile — classification and ADMET predicted profile — regression format. The prediction was displayed under three headings (model, result and probability) and the predicted model for absorption were Blood-Brain Barrier, Human Intestinal Absorption, Caco-2 Permeability, P-glycoprotein Substrate, P-glycoprotein Inhibitor and Renal Organic Cation Transporter; for distribution was Subcellular localization; metabolism were CYP450 2C9 Substrate, CYP450 2D6 Substrate, CYP450 3A4 Substrate, CYP450 1A2 Inhibitor, CYP450 2C9 Inhibitor, CYP450 2D6 Inhibitor, CYP450 2C19 Inhibitor, CYP450 3A4 Inhibitor and CYP Inhibitory Promiscuity and for toxicity, the Human Ether-a-go-go-Related Gene Inhibition, AMES Toxicity, Carcinogens, Fish Toxicity, Tetrahymena Pyriformis Toxicity, Honey Bee Toxicity, Biodegradation, Acute Oral Toxicity and Carcinogenicity (Three-class). Also, the factors considered for ADMET predicted profile — regression were Aqueous solubility, Caco-2 Permeability (absorption) and Rat Acute Toxicity, Fish Toxicity and Tetrahymena Pyriformis Toxicity (toxicity).Table 7. Pharmacokinetic evaluation of compound 10.Table 7:ADMET Predicted Profile — ClassificationModelResultProbabilityAbsorptionBlood-Brain BarrierBBB-0.96Human Intestinal AbsorptionHIA+0.98Caco-2 PermeabilityCaco2+0.88P-glycoprotein SubstrateSubstrate0.57P-glycoprotein InhibitorNon-inhibitor0.72Non-inhibitor0.74Renal Organic Cation TransporterNon-inhibitor0.94DistributionSubcellular localizationMitochondria0.44MetabolismCYP450 2C9 SubstrateSubstrate0.64CYP450 2D6 SubstrateNon-substrate0.91CYP450 3A4 SubstrateNon-substrate0.73CYP450 1A2 InhibitorNon-inhibitor0.90CYP450 2C9 InhibitorInhibitor0.90CYP450 2D6 InhibitorNon-inhibitor0.92CYP450 2C19 InhibitorNon-inhibitor0.90CYP450 3A4 InhibitorNon-inhibitor0.87CYP Inhibitory PromiscuityLow CYP Inhibitory Promiscuity0.87ToxicityHuman Ether-a-go-go-Related Gene InhibitionWeak inhibitor0.95Non-inhibitor0.83AMES ToxicityNon AMES toxic0.87CarcinogensNon-carcinogens0.76Fish ToxicityLow FHMT0.59Tetrahymena Pyriformis ToxicityHigh TPT0.93Honey Bee ToxicityLow HBT0.81BiodegradationNot ready biodegradable0.92Acute Oral ToxicityII0.72Carcinogenicity (Three-class)Non-required0.69ADMET Predicted Profile — RegressionModelValueUnitAbsorptionAqueous solubility-4.09LogSCaco-2 Permeability1.35LogPapp, cm/sToxicityRat Acute Toxicity3.15LD50, mol/kgFish Toxicity1.72pLC50, mg/LTetrahymena Pyriformis Toxicity0.53pIGC50, ug/LTable 8Pharmacokinetic evaluation of reference compound.Table 8:ADMET Predicted Profile — ClassificationModelResultProbabilityAbsorptionBlood-Brain BarrierBBB+0.96Human Intestinal AbsorptionHIA+0.99Caco-2 PermeabilityCaco2+0.88P-glycoprotein SubstrateNon-substrate0.75P-glycoprotein InhibitorNon-inhibitor0.97Non-inhibitor0.93Renal Organic Cation TransporterNon-inhibitor0.93DistributionSubcellular localizationMitochondria0.69MetabolismCYP450 2C9 SubstrateNon-substrate0.75CYP450 2D6 SubstrateNon-substrate0.91CYP450 3A4 SubstrateNon-substrate0.68CYP450 1A2 InhibitorNon-inhibitor0.90CYP450 2C9 InhibitorNon-inhibitor0.93CYP450 2D6 InhibitorNon-inhibitor0.92CYP450 2C19 InhibitorNon-inhibitor0.98CYP450 3A4 InhibitorNon-inhibitor0.96CYP Inhibitory PromiscuityLow CYP Inhibitory Promiscuity0.96ToxicityHuman Ether-a-go-go-Related Gene InhibitionWeak inhibitor0.97Non-inhibitor0.97AMES ToxicityNon AMES toxic0.98CarcinogensCarcinogens0.55Fish ToxicityHigh FHMT0.94Tetrahymena Pyriformis ToxicityHigh TPT0.99Honey Bee ToxicityHigh HBT0.74BiodegradationReady biodegradable0.51Acute Oral ToxicityIII0.80Carcinogenicity (Three-class)Non-required0.73ADMET Predicted Profile — RegressionModelValueUnitAbsorptionAqueous solubility-3.90LogSCaco-2 Permeability1.74LogPapp, cm/sToxicityRat Acute Toxicity2.30LD50, mol/kgFish Toxicity1.31pLC50, mg/LTetrahymena Pyriformis Toxicity1.38pIGC50, ug/L
Experimental Design, Materials and Methods
4
Ten heterocyclic compounds were modeled and optimized in three ways (selected compound; selected compound + Cadmium and selected compound + Lead) using Spartan ’14 [1,2]. These compounds were chosen based on their reported efficacy. Three selected features (HOMO, LUMO and energy gap) for each category for the entire compound under investigation were retrieved and reported. The entire compound were prepared for docking using induced fit docking method via molecular operating environment (MOE) software; while the receptor was retrieved from protein data bank Dipeptidyl Peptidase III [pdb id: 3fvy] [3] and treated by removing water molecules and small molecules (Zn, Mg and Cl) downloaded with the receptor and saved in moe format. The investigated complexes were calculated using induced fit docking method with 30 poses via molecular operating environment software [4,5]. The correlation between the calculated descriptors and the scoring for the examined complexes were executed using Microsoft excel software and the predicted squared correlation coefficient was reported accordingly. The pharmacokinetic evaluation of compound 10 and reference compound were accomplished using ADMETSar software [6,7] and the obtained outputs were reported.
Limitations
More investigation beyond what has been presented is expected so as to further probe the biological role of the investigated compounds.
Ethics Statement
This study does not involve studies with animals and humans.
CRediT authorship contribution statement
Kehinde Adeola Bolaji: Conceptualization, Methodology, Data curation, Writing – original draft, Visualization, Investigation, Writing – review & editing. Abel Kolawole Oyebamiji: Conceptualization, Methodology, Data curation, Writing – original draft, Visualization, Investigation, Writing – review & editing. Godwin Oladele Olutona: Conceptualization, Methodology, Data curation, Writing – original draft, Visualization, Investigation, Writing – review & editing.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Trejo-Muñoz C.R.Vázquez-Ramírez R.Mendoza L.Kubli-Garfias C.Biophysical mechanism of the SAHA inhibition of Zn 2+-histone deacetylase-like protein (FB 188 HDAH) assessed via crystal structure analysis Comput. Mol. Biosci.82201891114
- 2Adaramaja A.A.Bamisaye A.Abati S.M.Adegoke K.A.Adesina M.O.Ige A.R.Adeleke O.Idowu M.A.Oyebamiji A.K.Bello O.S.Thermally modified nanocrystalline snail shell adsorbent for methylene blue sequestration: equilibrium, kinetic, thermodynamic, artificial intelligence, and DFT studies RSC Adv.142024127033864552810.1039/d 4ra 01074 d PMC 11027000 · doi ↗ · pubmed ↗
- 3Bezerra G.A.Dobrovetsky E.Viertlmayr R.Dong A.Binter A.Abramic M.Macheroux P.Dhe-Paganon S.Gruber K.Entropy-driven binding of opioid peptides induces a large domain motion in human dipeptidyl peptidase III Proc. Natl. Acad. Sci. U. S. A.1092012652565302249323810.1073/pnas.1118005109 PMC 3340026 · doi ↗ · pubmed ↗
- 4Binjawhar D.N.Ali O.A.A Alqahtani A.S.Fayad E.Abo-Bakr A.M.Mekhael A.M.Sadek F.M.Powerful approach for new drugs as antibacterial agents via molecular docking and invitro studies of some new cyclic imides and quinazoline-2,5-diones ACS Omega 916202418566185753868034010.1021/acsomega.4c 01176 PMC 11044208 · doi ↗ · pubmed ↗
- 5Shen J.Cheng F.Xu Y.Li W.Tang Y.Estimation of ADME properties with substructure pattern recognition J. Chem. Inf. Model.5062010103410412057872710.1021/ci 100104 j · doi ↗ · pubmed ↗
- 6Gu Y.Wang Y.Zhu K.DBPP-predictor: a novel strategy for prediction of chemical drug-likeness based on property profiles J. Cheminform.162024410.1186/s 13321-024-00800-938183072 PMC 10771006 · doi ↗ · pubmed ↗
- 7Wang Y.Huang M.Deng H.Li W.Wu Z.Tang Y.Liu G.Identification of vital chemical information via visualization of graph neural networks Brief. Bioinform.2412023 bbac 57710.1093/bib/bbac 57736537081 · doi ↗ · pubmed ↗