Dataset on potential biochemical activities of Cylo phe-Ala-Asp-Gly-based compounds as Caspase 1 inhibitor
Faith Eniola Olujinmi, Chijioke John Ajaelu, Sunday Adewale Akintelu, Abel Kolawole Oyebamiji

TL;DR
This study investigates seven compounds as potential inhibitors of human Caspase 1, identifying one as particularly effective.
Contribution
A new compound is identified as a more efficient Caspase 1 inhibitor compared to others.
Findings
Compound 2 showed higher inhibiting efficiency against human Caspase 1.
Pharmacokinetic evaluations were performed on the most effective compound and a reference compound.
Molecular docking and descriptor calculations were used to assess biochemical activities.
Abstract
In this work, seven Cylo phe-Ala-Asp-Gly-based Compounds were optimized using Spartan’14 software. Series of software used in this work were Spartan 14, molecular operating environment (MOE), padel, ADMET. The optimized compounds were docked against human Caspase 1 (6F6R) so as to observe their inhibiting ability. The calculated descriptors for individual compounds were reported and described. Also, the docked compound 2 (2-((2S,8S,11S)-11-([1,1′-biphenyl]-4-ylmethyl)-8-benzyl-3,6,9,12-tetraoxo-1,4,7,10-tetraazacyclododecan-2-yl) acetic acid) proved to be more efficient in inhibiting human caspase 1 than other compounds under investigation. The pharmacokinetic evaluation of compound with highest binding affinity and reference compound were executed and reported.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
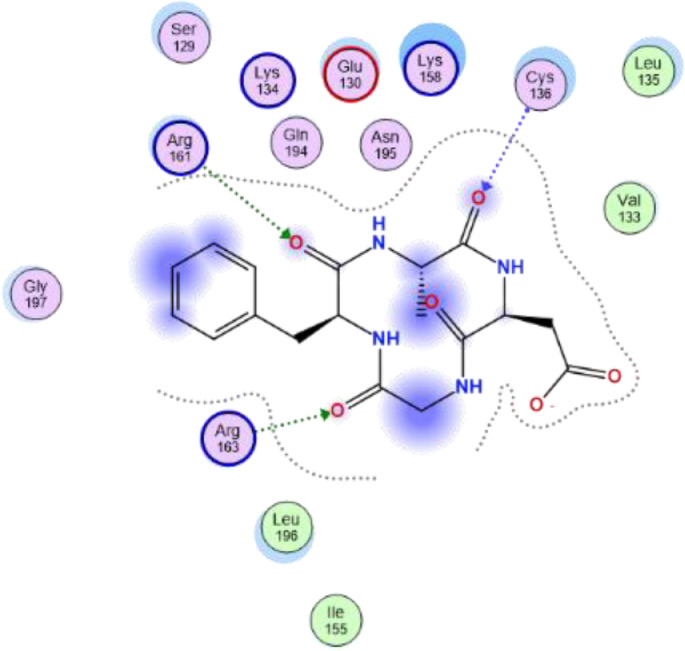 Figure 1
Figure 1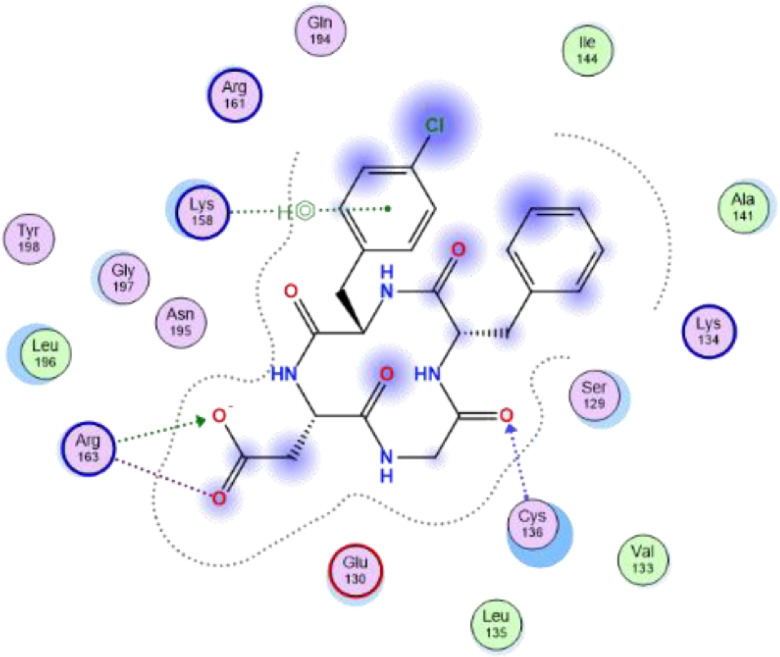 Figure 2
Figure 2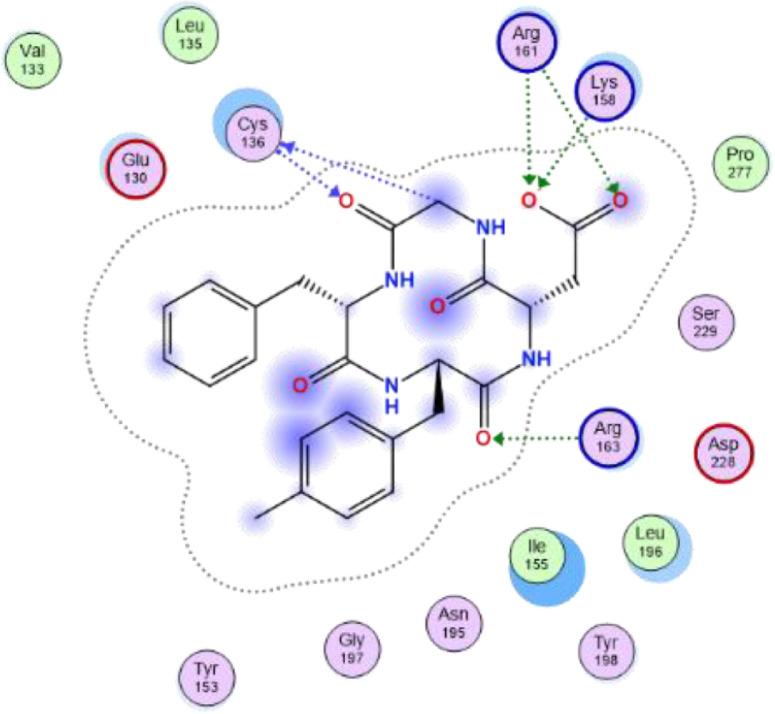 Figure 3
Figure 3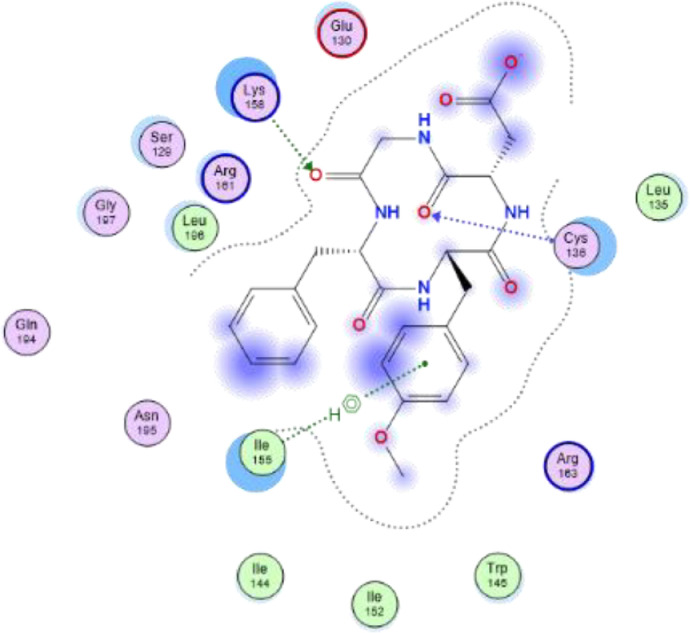 Figure 4
Figure 4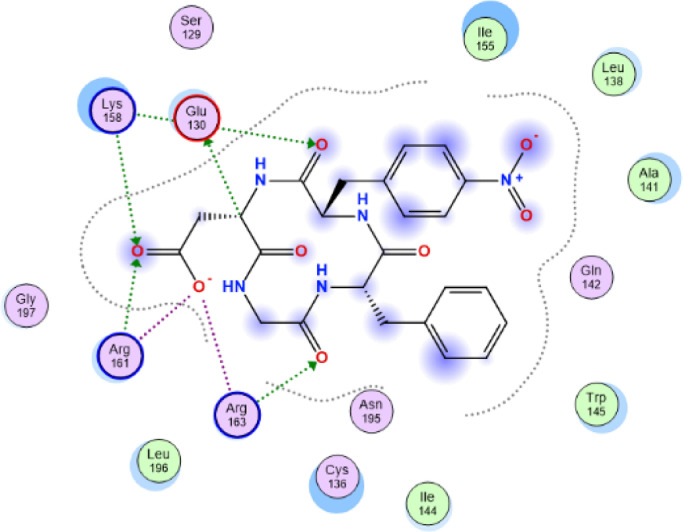 Figure 5
Figure 5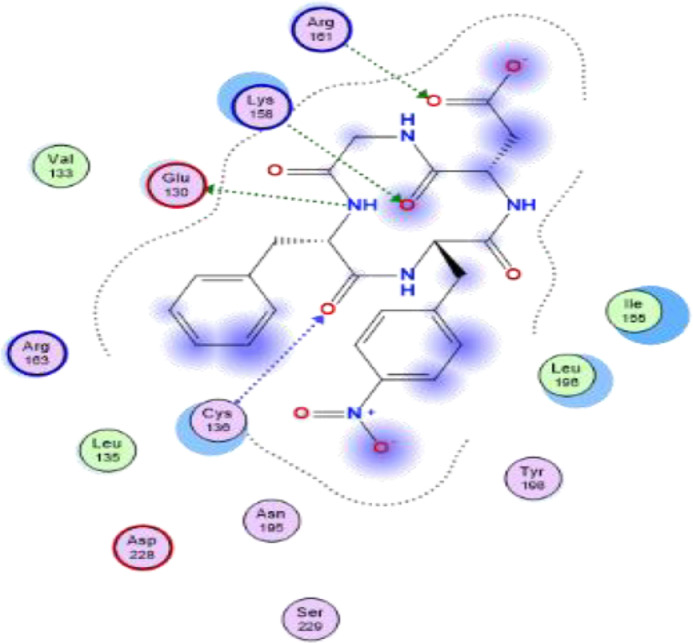 Figure 6
Figure 6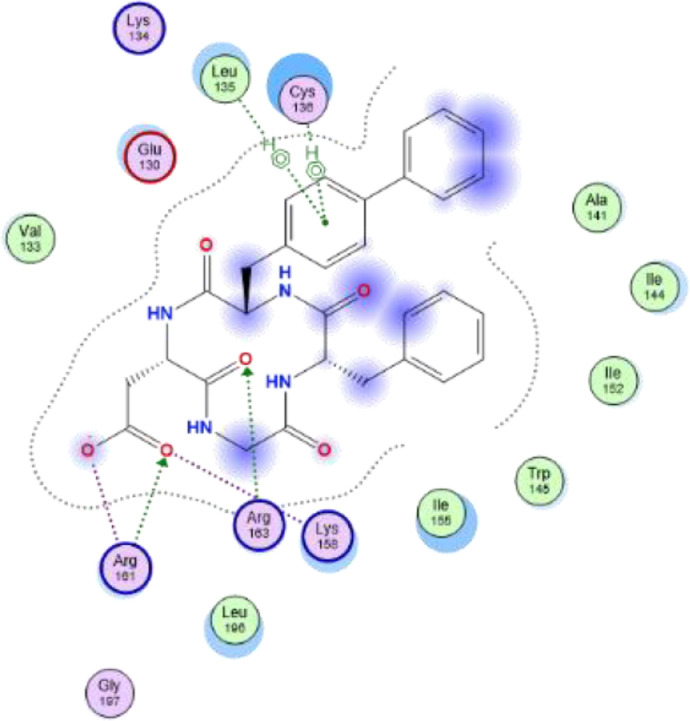 Figure 7
Figure 7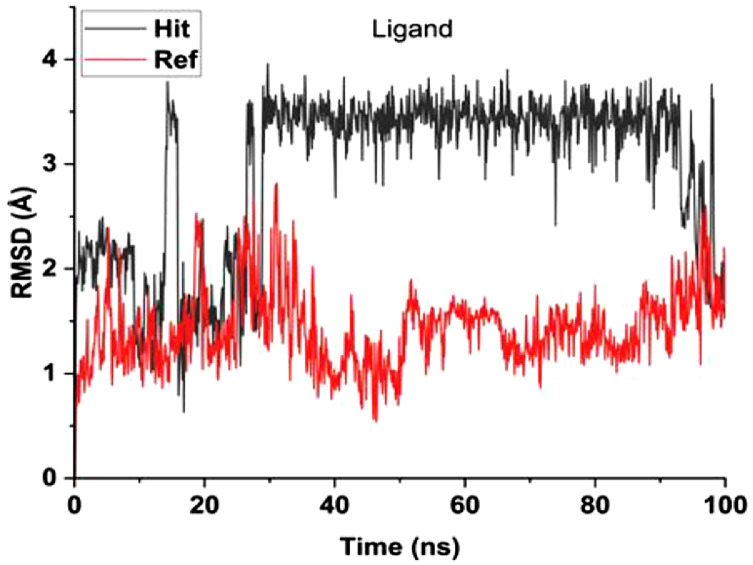 Figure 8
Figure 8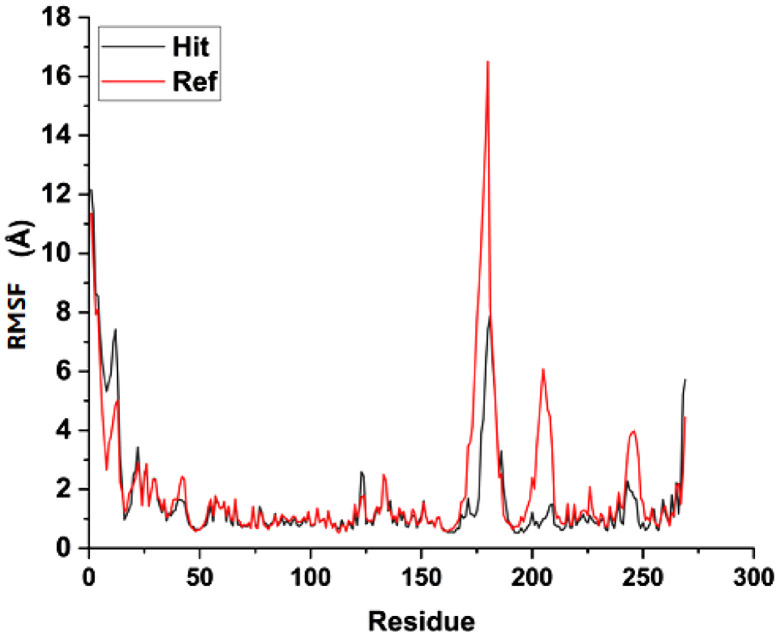 Figure 9
Figure 9Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsComputational Drug Discovery Methods · ATP Synthase and ATPases Research · Machine Learning in Bioinformatics
Specifications TableSubjectComputational Chemistry.Specific subject areaDrug Design and AnalysisType of dataRaw Data, Figure, Chart, Scheme, Table, ADMET, GraphData collectionSeven Cylo phe-Ala-Asp-Gly-based Compounds were modelled and subjected to density functional theory calculation via B3LYP (6-31+G*) for theoretical calculations using Spartan ‘14. The compounds were calculated in three phases (gas, ethanol and water) and the descriptors were presented accordingly. The docking calculation was accomplished using induced fit method for molecular docking investigation. The pharmacokinetic evaluation of the compound with best binding affinity was executed using ADMETSar 2.0 and all the results were reported appropriately.Data source locationComputational Chemistry Research Laboratory, Department of Chemistry and Industrial Chemistry, BOWEN University, PMB 284, Iwo, Osun State, Nigeria.Data accessibility***Please note:*All raw data referred to in this article must be made publicly available in a data repository prior to publication. Please indicate here where your data are hosted (the URL must be working at the time of submission and editors and reviewers must have anonymous access to the repository):Repository name: Mendeley DataData identification number: doi:10.17632/hn36p8mp4r.1Direct URL to data: https://data.mendeley.com/datasets/hn36p8mp4r/1Instructions for accessing these data: The data can be accessed using the above URL.Related research articleNone.
Value of the Data
1
- •The molecular descriptors obtained from the optimized compound in the three different phases will provide information to researchers (most especially, organic Chemist) on the effect of solvent and most suitable solvent for the compounds.
- •The calculated binding affinity for the optimized compound against (PDB ID: 6F6R) Caspase-1 will help scientists (especially, pharmaceutical scientists/ medicinal Chemist) to identify compounds that possess the best configuration with utmost efficiency.
- •The 2- dimensional structures of the compounds under investigation will reveal to researchers the nature of the derivatives attached to the parent compound.
- •The distance between every atom in the compounds and the amino acid residues will expose to scientists the level of spontaneity of the examined peptide-based compound and receptor.
- •The pharmacokinetic analysis will provide insight into how the compound with the best binding affinity is absorbed, digested, metabolized and excreted in the body and also reveal the toxicity level.
- •The molecular dynamic simulation will help researchers to confirm the ligand with utmost potential anti- Caspase 1 properties.
Background
2
The objectives of this work are:
- ➢To evaluate solvent effect on the chemical activity of the examined Cylo phe-Ala-Asp-Gly-based Compounds
- ➢To observe the interaction between the compound and the amino acid residues in the active site of Caspase-1 (PDB ID: 6F6R)
- ➢To reveal the pharmacokinetics activity of the compound with the best binding affinity*.*
- ➢To identify the molecule with highest potential capacity to inhibit Caspase 1 which will thereby help in predicting libraries of efficient compounds
Data Description
3
Table 1 reveals the seven compounds which are all derivatives of the parent compound: Cylo phe-Ala-Asp-Gly, the IUPAC nomenclature and the two-dimensional structure of the compounds investigated in this work are displayed in Table 1.Table 1. Two-dimensional structure of the examined peptide based compounds.Table 1
Table 2 exposes the descriptors obtained for the investigated compound. The highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital (LUMO) and the energy gap in all three phases are detailed in Table 2.The location of the orbital energy levels HOMO-LUMO for the seven compounds being examined are listed in Table 3. HOMO-LUMO was obtained for all the optimised compounds in the vacuum, water and ethanol using dot format for the three phases (Table 4, Table 5).Table 2. Calculated DESCRIPTORS for compounds optimized in vacuum, water and ethanol.Table 2. VacuumWaterEthanolHOMOLUMOEGHOMOLUMOEGHOMOLUMOEG1-7.05-1.34-5.71-6.91-1.37-5.54-6.82-1.42-5.42-6.71-1.57-5.14-6.8-1.36-5.44-6.77-1.29-5.483-6.75-1.52-5.23-6.55-1.44-5.11-6.56-1.38-5.184-6.72-1.52-5.2-6.17-1.4-4.77-6.16-1.34-4.825-7.1-3.78-3.32-6.91-4.22-2.69-6.84-3.89-2.956-6.68-1.57-5.11-6.76-1.37-5.39-6.74-1.3-5.447-6.75-1.56-5.19-6.45-1.44-5.01-6.46-1.38-5.08Table 3Predicted orbital energy levels for compounds optimized in vacuum.Table 3. Table 4Predicted orbital energy levels for compounds optimized in water.Table 4
Table 6 reveals the binding affinity of the Cylo phe-Ala-Asp-Gly compounds against the human caspase 1 enzyme (PDB ID: 6F6R). The binding affinity is -6.5045414 kcal/mol for compound 1, -6.9591408 kcal/mol for compound 2, -7.248467 kcal/mol for compound 3, -6.9861174 kcal/mol compound 4, -7.0307384 kcal/mol for compound 5 -6.8699188 for compound 6, -7.4287872 kcal/mol and the reference drug -7.4287872 kcal/mol with which all the compound was compared.
Table 7 reveals the ligand and amino acid residue involved in the interaction between the compounds and the human caspase 1 (6F6R). It also shows the distance between the ligand and the active site of the receptor. The two-dimensional structure scheme showing the interaction between each investigated compound and the proteins in the active site human caspase 1 receptor (6F6R) is displayed in Fig. 1, Fig. 2, Fig. 3, Fig. 4, Fig. 5, Fig. 6, Fig. 7–8.Fig. 12-dimensional structure for docked Compound 1 against human Caspase 1 [pdb id: 6F6R].Fig 1. Fig. 22-dimensional structure for docked Compound 2 against human Caspase 1 [pdb id: 6F6R].Fig 2. Fig. 32-dimensional structure for docked Compound 3 against human Caspase 1 [pdb id: 6F6R].Fig 3. Fig. 42-dimensional structure for docked Compound 4 against human Caspase 1 [pdb id: 6F6R].Fig 4. Fig. 52-dimensional structure for docked Compound 5 against human Caspase 1 [pdb id: 6F6R].Fig 5. Fig. 62-dimensional structure for docked Compound 6 against human Caspase 1 [pdb id: 6F6R].Fig 6. Fig. 72-dimensional structure for docked Compound 7 against human Caspase 1 [pdb id: 6F6R].Fig 7. Table 5Predicted orbital energy levels for compounds optimized in ethanol.Table 5. Table 6Calculated binding affinity for examined complexes.Table 6. Scoring (kcal/mol)1-6.50454142-6.95914083-7.2484674-6.98611745-7.03073846-6.86991887-7.4287872VX-740-6.7491722Table 7Predicted non bonding interaction for examined complexes.Table 7. LigandReceptorInteractionDistanceE(Kcal/mol)1O2 2NH2 ARG 161 (A)H-acceptor3.52-0.7O13 13N CYS 136 (A)H-acceptor3.37-0.8O26 26NH1 ARG 163 (A)H-acceptor3.07-1.152O24 24NH1 ARG 163 (A)H-acceptor2.96-3.6O24 24NH2 ARG 163 (A)H-acceptor3.22-1.7O26 26N CYS 136 (A)H-acceptor2.94-1.3O23 23NH1 ARG 163 (A)Ionic3.01-4.4O23 23NH2 ARG 163 (A)Ionic3.8-1O24 24NH1 ARG 163 (A)Ionic2.96-4.7O24 24NH2 ARG 163 (A)Ionic3.22-3.26-ringCE LYS 158pi-H4.48-0.53C28 28O CYS 136 (A)H-donor3.46-0.7O13 13NH2 ARG 163 (A)H-acceptor3.16-2.9O23 23NH1 ARG 161 (A)H-acceptor2.96-2.5O24 24CD LYS 158 (A)H-acceptor3.02-0.6O24 24NH2 ARG 161 (A)H-acceptor2.96-2.2O26 26N CYS 136 (A)H-acceptor2.9-1.6O23 23NH1 ARG 161 (A)Ionic2.96-4.8O23 23NH2 ARG 161 (A)Ionic3.74-1.1O24 24NH1 ARG 161 (A)Ionic3.66-1.3O24 24NH2 ARG 161 (A)Ionic2.964.74O18 18N CYS 136 (A)H-acceptor2.94-3.3026 26NZ LYS 158 (A)H-acceptor2.54-4.66-ringCG2 ILE 155 (A)pi-H3.77-0.75C20 20OE2 GLU 130 (A)H-donor3-1O13 13NZ LYS 158 (A)H-acceptor3.22-2.2O23 23CE LYS 158 (A)H-acceptor3.51-0.6O23 23NH2 ARG 161 (A)H-acceptor2.78-0.7O26 26NH1 ARG 163 (A)H-acceptor3.47-1.7O23 23NH2 ARG 161 (A)Ionic2.78-6.2O24 24NH2 ARG 161 (A)Ionic2.65-7.3O24 24NH1 ARG 163 (A)Ionic3.25-36N3 3OE2 GLU 130 (A)H-donor3.47-0.6O2 2N CYS 136 (A)H-acceptor3-4.4O18 18NZ LYS 158 (A)H-acceptor3.16-6.2O23 23NH1 ARG 161 (A)H-acceptor3.05-9.4O23 23NH1 ARG 161 (A)Ionic3.05-4.1O23 23NH2 ARG 161 (A)Ionic3.8-17O18 18NH1 ARG 163 (A)H-acceptor3.19-2.6O18 18NH2 ARG 163 (A)H-acceptor3.26-1.7O23 23NH2 ARG 161 (A)H-acceptor3.12-1.3O23 23NZ LYS 158 (A)Ionic3.82-0.9O23 23NH2 ARG 161 (A)Ionic3.12-3.7O24 24NH2 ARG 161 (A)Ionic2.95-4.86-ringCA LEU 135 (A)pi-H4.18-0.86-ringN CYS 136 (A)pi-H3.62-0.8Fig. 8RMSD for compound 7 and VX-740 against human Caspase 1 (6F6R).Note: Hit: Compound 7, Ref: VX-740.Fig 8
Table 8, Table 9 shows the result from the pharmacokinetic study of the compound with the best binding affinity (Compound 7) and the reference compound. The result was presented in ADMET-predicted profile classification and ADMET-predicted profile regression format. The predictions for the absorption model were Blood–Brain Barrier, Human Intestinal Absorption, Caco-2 Permeability, P-glycoprotein Substrate, P-glycoprotein Inhibitor and Renal Organic Cation Transporter. The distribution model is Subcellular localization, metabolism model was CYP450 2C9 Substrate, CYP450 2D6 Substrate, CYP450 3A4 Substrate, CYP450 1A2 Inhibitor, CYP450 2C9 Inhibitor, CYP450 2D6 Inhibitor, CYP450 2C19 Inhibitor, CYP450 3A4 Inhibitor and CYP Inhibitory Promiscuity and toxicity model: the Human Ether-a-go-go-Related Gene Inhibition, AMES Toxicity, Carcinogens, Fish Toxicity, Tetrahymena Pyriformis Toxicity, Honey Bee Toxicity, Biodegradation, Acute Oral Toxicity and Carcinogenicity (Three-class). For ADMET profiling prediction the following regression factors were considered aqueous solubility, Caco-2 Permeability (absorption) and Rat Acute Toxicity, Fish Toxicity and Tetrahymena Pyriformis Toxicity (toxicity).Table 8. Predicted pharmacokinetic evaluation for compound 7.Table 8ADMET Predicted Profile — ClassificationModelResultProbabilityAbsorptionBlood-Brain BarrierBBB-0.6541Human Intestinal AbsorptionHIA±0.8887Caco-2 PermeabilityCaco2-0.7925P-glycoprotein SubstrateSubstrate0.6391P-glycoprotein InhibitorNon-inhibitor0.9087Non-inhibitor0.9682Renal Organic Cation TransporterNon-inhibitor0.9231DistributionSubcellular localizationMitochondria0.6841MetabolismCYP450 2C9 SubstrateNon-substrate0.8071CYP450 2D6 SubstrateNon-substrate0.8191CYP450 3A4 SubstrateNon-substrate0.6056CYP450 1A2 InhibitorNon-inhibitor0.8966CYP450 2C9 InhibitorNon-inhibitor0.9276CYP450 2D6 InhibitorNon-inhibitor0.9467CYP450 2C19 InhibitorNon-inhibitor0.9347CYP450 3A4 InhibitorNon-inhibitor0.9231CYP Inhibitory PromiscuityLow CYP Inhibitory Promiscuity0.9536Excretion****ToxicityHuman Ether-a-go-go-Related Gene InhibitionWeak inhibitor0.9903Non-inhibitor0.8005AMES ToxicityNon AMES toxic0.8748CarcinogensNon-carcinogens0.9227Fish ToxicityHigh FHMT0.9579Tetrahymena Pyriformis ToxicityHigh TPT0.9027Honey Bee ToxicityLow HBT0.8209BiodegradationNot ready biodegradable0.9959Acute Oral ToxicityIII0.6314Carcinogenicity (Three-class)Non-required0.7671ADMET Predicted Profile — RegressionModelValueUnit****AbsorptionAqueous solubility-3.0091LogSCaco-2 Permeability-0.4886LogPapp, cm/sDistributionMetabolismExcretion****ToxicityRat Acute Toxicity2.3315LD50, mol/kgFish Toxicity1.8950pLC50, mg/LTetrahymena Pyriformis Toxicity0.0777pIGC50, ug/LTable 9Predicted pharmacokinetic evaluation for reference compound.Table 9ADMET Predicted Profile — ClassificationModelResultProbabilityAbsorptionBlood-Brain BarrierBBB-0.8386Human Intestinal AbsorptionHIA±0.9821Caco-2 PermeabilityCaco2-0.7237P-glycoprotein SubstrateSubstrate0.7204P-glycoprotein InhibitorInhibitor0.7443Non-inhibitor0.9365Renal Organic Cation TransporterNon-inhibitor0.7706DistributionSubcellular localizationMitochondria0.5360MetabolismCYP450 2C9 SubstrateNon-substrate0.8340CYP450 2D6 SubstrateNon-substrate0.8350CYP450 3A4 SubstrateSubstrate0.6186CYP450 1A2 InhibitorNon-inhibitor0.8814CYP450 2C9 InhibitorNon-inhibitor0.7453CYP450 2D6 InhibitorNon-inhibitor0.8095CYP450 2C19 InhibitorNon-inhibitor0.7977CYP450 3A4 InhibitorNon-inhibitor0.5643CYP Inhibitory PromiscuityHigh CYP Inhibitory Promiscuity0.5861Excretion****ToxicityHuman Ether-a-go-go-Related Gene InhibitionWeak inhibitor0.9197Non-inhibitor0.5157AMES ToxicityNon AMES toxic0.5288CarcinogensNon-carcinogens0.8882Fish ToxicityHigh FHMT0.9994Tetrahymena Pyriformis ToxicityHigh TPT0.9617Honey Bee ToxicityLow HBT0.8033BiodegradationNot ready biodegradable1.0000Acute Oral ToxicityIII0.6686Carcinogenicity (Three-class)Non-required0.6185ADMET Predicted Profile — RegressionModelValueUnit****AbsorptionAqueous solubility-3.2289LogSCaco-2 Permeability0.3905LogPapp, cm/sDistributionMetabolismExcretion****ToxicityRat Acute Toxicity2.4106LD50, mol/kgFish Toxicity1.3558pLC50, mg/LTetrahymena Pyriformis Toxicity0.4015pIGC50, ug/L
Table 10 showed the calculated binding energy between the lead compound and the reference compound. The free energy calculated in this work was ΔG_gas_ (gas phase energy), ΔG_solv_ (solvation free energy) and ΔG_bind_ (binding free energy). The calculated gas phase energy, solvation free energy and binding free energy for compound 7 were -37.4808 kcal/mol, 20.6774 kcal/mol and -16.8034 kcal/mol. More so, the gas phase energy, solvation free energy and binding free energy calculated for the reference compound were -61.5856 kcal/mol, 18.5007 kcal/mol and -15.0850 kcal/mol. Also, Fig. 8, Fig. 9 revealed the root mean square deviation (RMSD) carried out in 100ns and root mean square fluctuation (RMSF) respectively. The lead compound was represented in black line while the reference compound was represented in red line.Table 10. Calculated binding energy for compound 7-6F6R and Ref-6F6R.Table 10. ComplexΔG_gas_ΔG_solv_ΔG_bind_ (kcal/mol)Hit-37.480820.6774-16.8034Reference-61.585618.5007-15.0850Note: Hit: Compound 7, Reference: Vx-740Fig. 9RMSF for compound 7 and VX-740 against human Caspase 1 (6F6R).Note: Hit: Compound 7, Ref: VX-740.Fig 9
Experimental Design, Materials and Methods
4
The 2-dimensional structures of the compounds under investigations were modelled using Chemdraw software and further subjected to Spartan 14 software which thereby convert the compounds to 3-dimensional structure. Spartan 14 software was used to optimize the Cylo phe-Ala-Asp-Gly compounds using density functional theory methodology via 6-31+ G* as basis set for the entire compounds which were optimized three phases: vacuum, water and ethanol. Optimization of the compounds in these three phases generated various molecular descriptors which describes the drug like activities of the compounds under investigation [1]. Also, human caspase 1 enzyme with pdb id: 6F6R [2] was retrieved from the protein data bank and the enzyme was treated using Molecular operating environment software to get rid of water molecules, and other small molecules before subjecting to docking calculation [[3], [4], [5]]. The small molecule retrieved from the receptor was docked into the active site of the receptor and the docked retrieved compound was placed on the original retrieved compound using discovery studio software to discern the level of deviation. It was observed that the RMSD was closer to 1 which confirmed the reliability of docking method used in this work. The examined enzyme structure was optimized using quickprep tool in order to repair and prepare the receptor before docking calculation. Human capase-1 enzyme was subjected to molecular operating environment to identify the potential binding sites the the Cylo phe-Ala-Asp-Gly compounds will bind with. Scoring of the calculated binding affinity measure in kcal/mol was also obtained and the result was accurately presented.
The interaction between compound 7 as well as the reference compound and the caspase-1 enzyme (PDB ID: 6F6R) for 100 ns were examined using molecular dynamics (MD).The simulated system was solvated with water molecules and the addition of counterions to neutralize the system. The CHARMM force field was typically chosen to model the interactions between the enzyme, compound, water, and ions. An initial energy minimization was performed to relieve any steric clashes, followed by equilibration in the NPT ensemble to stabilize temperature and pressure. The system was then subjected to a 100 ns production run, with periodic updates of atom positions and velocities using an integration time step of 1-2 fs. Throughout the simulation, key properties such as the binding stability, conformational changes of caspase-1, and interactions with compound 7 as well as the reference compound were monitored. The trajectory data were then analyzed to gain insights into binding affinities, molecular interactions, and dynamics of the enzyme-ligand complex [[6], [7], [8], [9]].
Limitations
Not applicable
Ethics Statement
This study does not involve studies with animals and humans*.*
CRediT authorship contribution statement
Faith Eniola Olujinmi: Conceptualization, Methodology, Data curation, Writing – original draft, Visualization, Investigation, Writing – review & editing. Chijioke John Ajaelu: Conceptualization, Methodology. Sunday Adewale Akintelu: Data curation, Writing – original draft, Visualization, Investigation, Writing – review & editing. Abel Kolawole Oyebamiji: Conceptualization, Methodology, Data curation, Writing – original draft, Visualization, Investigation, Writing – review & editing.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Oyebamiji A.K.Akintelu S.A.Akintayo E.T.Akintayo C.O.Aworinde H.O.Adekunle O.D.Dataset on substituents effect on biological activities of linear RGD-containing peptides as potential anti-angiotensin converting enzyme Data Br.50202310947810.1016/j.dib.2023.109478 PMC 1043260637600591 · doi ↗ · pubmed ↗
- 2Archana C.M.Harini K.Showmya J.J.Geetha N.An in-silico docking study of chromolaena odorata derived compounds against antimalarial activity Int. J. Pharmacol. Sci. Bus. Manag.2920144258
- 3Oyebamiji A.K.Olujinmi F.E.Aworinde H.O.Oke D.G.Akintelu S.A.Akintayo E.T.Akintayo C.O.Babalola J.O.Dataset on anti-human insulin-degrading enzyme activities of cyclic tetra peptides: insight from in silico approach Data Br.202411072410.1016/j.dib.2024.110724 PMC 1129554239100774 · doi ↗ · pubmed ↗
- 4Attique S.A.Hassan M.Usman M.Atif R.M.Mahboob S.Al-Ghanim K.A.Bilal M.Nawaz M.Z.A molecular docking approach to evaluate the pharmacological properties of natural and synthetic treatment candidates for use against hypertension Int. J. Environ. Res. Public Health 16620199233087581710.3390/ijerph 16060923 PMC 6466102 · doi ↗ · pubmed ↗
- 5Mahmoud N.F.Abbas A.A.Mohamed G.G.Synthesis, characterization, antimicrobial, and MOE evaluation of nano 1, 2, 4-triazole-based Schiff base ligand with some d-block metal ions Appl. Organomet. Chem.3562021 e 6219
- 6Wei H.Mc Cammon J.A.Structure and dynamics in drug discoverynpj Drug Discov.11202410.1038/s 44386-024-00001-2 · doi ↗
- 7Bhati S.K.Anjum F.Shamsi A.In silico screening and molecular dynamics analysis of natural DHPS enzyme inhibitors targeting Acinetobacter baumannii Sci. Rep.152025772310.1038/s 41598-025-90946-940044750 PMC 11883060 · doi ↗ · pubmed ↗
- 8Zhen H.Hu Y.Liu X.Fan G.Zhao S.The protease caspase-1: activation pathways and functions Biochem. Biophys. Res. Commun.717202414997810.1016/j.bbrc.2024.14997838718564 · doi ↗ · pubmed ↗