General Synthesis of 2-Substituted Benzoxazoles Based on Tf2O-Promoted Electrophilic Activation of Tertiary Amides
Hongchen Li, Xingyong Wang, Fujun Zhao, Lu Wang, Songbao Fu

TL;DR
A new method is introduced for making 2-substituted benzoxazoles using triflic anhydride and 2-fluoropyridine, enabling versatile synthesis of various derivatives.
Contribution
A novel Tf2O-promoted cascade reaction for synthesizing 2-substituted benzoxazoles from tertiary amides and 2-aminophenols.
Findings
The method involves activation of amide carbonyl groups by Tf2O followed by nucleophilic addition and cyclization.
The reaction is versatile, allowing synthesis of diverse benzoxazole derivatives by varying substrates.
The cascade process includes intramolecular cyclization and elimination steps.
Abstract
We report a method for the synthesis of 2-substituted benzoxazoles from tertiary amides and 2-aminophenols in the presence of triflic anhydride (Tf2O) and 2-Fluoropyridine (2-F-Pyr). The cascade reaction involves the activation of the amide carbonyl group by Tf2O, nucleophilic addition, intramolecular cyclization, and elimination. Furthermore, we explore the scope of this method by varying both the amide and 2-aminophenol substrates, highlighting its versatility in the synthesis of a wide range of functionalized benzoxazole derivatives.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
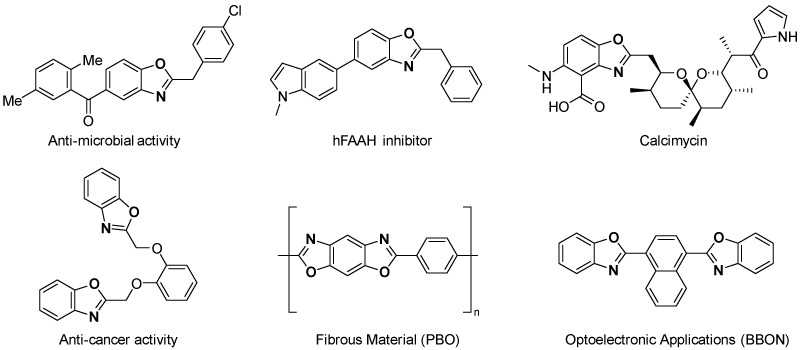 Figure 1
Figure 1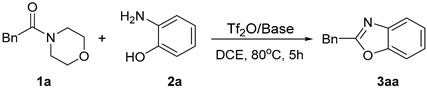 Figure 2
Figure 2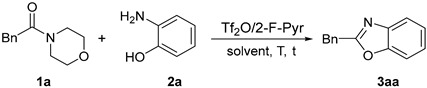 Figure 3
Figure 3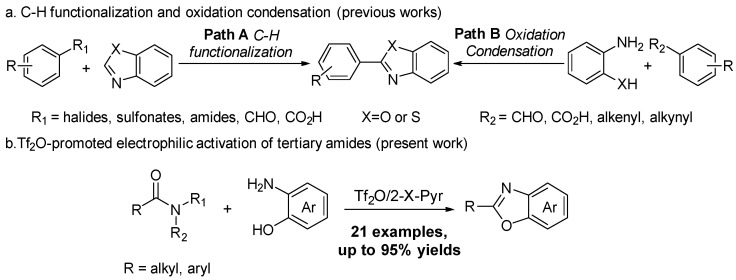 Figure 4
Figure 4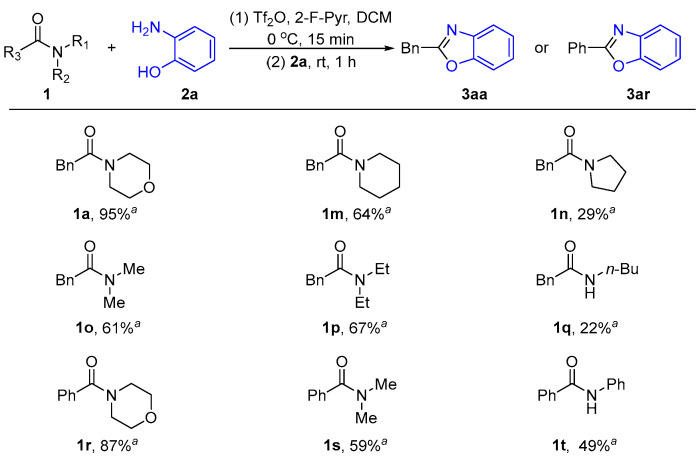 Figure 5
Figure 5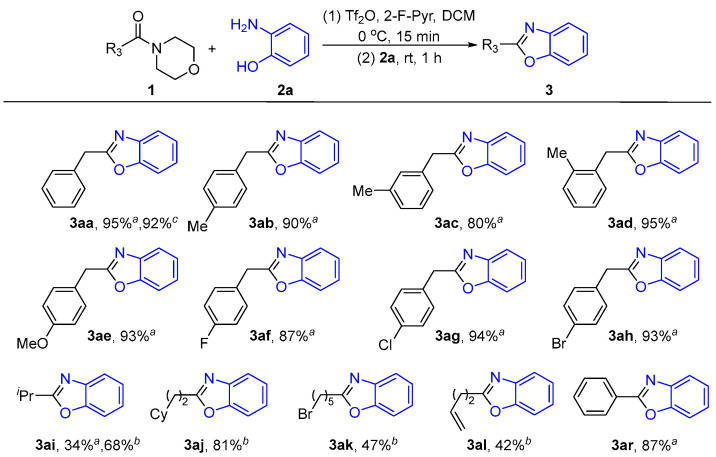 Figure 6
Figure 6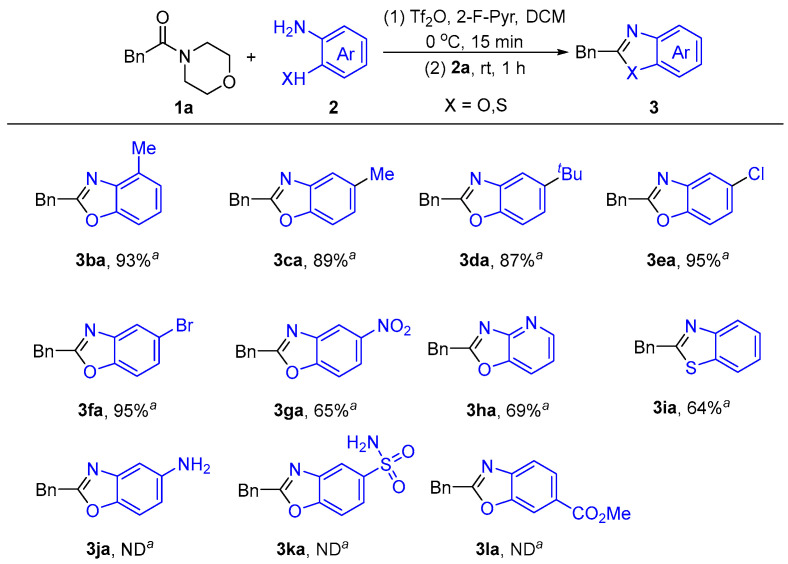 Figure 7
Figure 7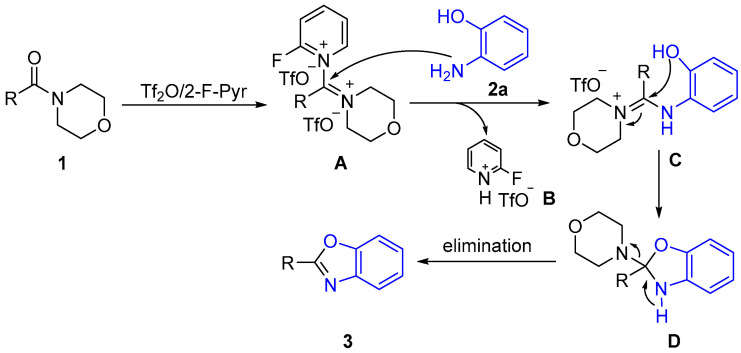 Figure 8
Figure 8- —the National Natural Science Foundation of China
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAdvanced Synthetic Organic Chemistry · Chemical Synthesis and Analysis · Chemical synthesis and alkaloids
1. Introduction
The 2-substituted benzoxazoles are an important class of heterocyclic compounds [1,2,3,4]. These compounds have attracted significant attention for their promising applications in medicinal chemistry [5,6,7,8,9,10,11,12,13,14,15,16,17], materials science [18], and organic electronics [19]. In particular, 2-substituted benzoxazoles exhibit a range of pharmacological activities, including anti-microbial [6], anti-cancer [8], anti-inflammatory [16], and anticonvulsant effects [17], which makes them valuable scaffolds for the development of new therapeutic agents (Figure 1).
Given the importance of 2-substituted benzoxazoles, numerous synthetic strategies have been developed to construct these valuable heterocyclic frameworks. Currently, the synthesis of 2-substituted benzoxazoles can be broadly categorized into two main approaches (Scheme 1a) [20,21,22]. The first approach involves the direct functionalization of the C-H bond at the C2 position, which allows the direct functionalization of the benzoxazole core (Scheme 1a, Path A) [23,24,25,26]. This strategy has attracted considerable attention due to its high atom economy and the ability to introduce a wide variety of functional groups without the need for pre-functionalized substrates. However, many of these transformations still require strong oxidants, high temperatures or very low temperatures, and special catalysts, which can limit their practical application. In addition, the use of transition metal catalysts poses challenges in pharmaceutical and industrial applications due to the presence of metal residues that require additional purification steps.
In view of the challenges associated with direct C-H functionalization at the C2 position of benzoxazoles, an alternative and widely used strategy for the synthesis of 2-substituted benzoxazoles is the cyclization of suitably functionalized precursors [27,28,29,30,31,32,33]. This method makes it possible to construct the benzoxazole core straightforwardly and to introduce the desired substituent at the C2 position at the same time (Scheme 1a, Path B). One of the most common methods is the cyclization of 2-aminophenols with aryl or alkyl precursors, such as benzoic acids [27], aryl methyl ketones [28,29], alkynes [30], and styrene derivatives [31,32,33]. These cyclization strategies provide a more general and versatile route to 2-substituted benzoxazoles. They often show high selectivity and better substrate tolerance compared to C-H activation methods.
In recent years, amide transformation has gained significant attention as a starting material for the synthesis of heterocyclic compounds [34,35,36,37,38]. This is due to the availability and versatility of amide substrates. However, efficient activation of the amide functional group is the key challenge in using amides for heterocycle synthesis. Amides are generally considered to be relatively inert compared to other carbonyl-containing compounds, such as esters or acids. This is mainly because the amide is affected by the increased stability of the electrophilic carbonyl carbon, resulting in much lower reactivity with nucleophilic reagents [34]. Under mild conditions, this makes direct reactions difficult. Recent advances in electrophilic activation strategies, including using strong acids like Tf_2_O, have proven to be very effective [39,40,41,42,43,44]. Herein, we report a method that explores the use of Tf_2_O-promoted electrophilic activation to synthesize 2-substituted benzoxazoles, highlighting its potential as a more selective and efficient activation strategy compared to traditional methods (Scheme 1b).
The ability of Tf_2_O to activate amides through the formation of highly reactive intermediates opens up new ways of functionalizing both amides and 2-aminophenols. This provides access to a wide range of substituted benzoxazoles.
2. Results and Discussion
2.1. Synthesis
2.1.1. Optimization of Reaction Conditions
As part of our continuing research into amide activation to construct heterocyclic compounds, we hypothesized that amides could be directly cyclized as starting materials to synthesize 2-substituted benzoxazoles. As expected, we successfully obtained the target product 2-benzylbenzo[d]oxazole (3aa) by the reaction of 1-morpholino-2-phenylethan-1-one (1a) and 2-aminophenol (2a) in the presence of Tf_2_O (Table 1, entry 1). Based on these preliminary results, we investigated different reaction conditions to optimize the process. First, the effect of different bases on the reaction was tested. The results are summarized in Table 1. The yield of the product was significantly increased by adding pyridine-based organic bases to the reaction (Table 1, entries 2-9). However, if the pyridine ring contains strong electron-absorbing functional groups (such as –SO_3_H and –NO_2_), the effect of the pyridine base is less obvious and the yield is reduced (Table 1, entries 8-9). When inorganic bases, such as CsF and K_2_CO_3_, were used, the reaction showed little improvement (Table 1, entries 10-11). The reaction yield was significantly increased by increasing the amount of 2-Fluoropyridine (Table 1, entries 12–14).
We then further optimized the reaction by exploring the impact of different conditions. A variety of solvents were tested, and DCM gave the best results (Table 2, entries 1–4). Reducing the reaction temperature had little effect on the yield, with the highest yield being obtained at room temperature (Table 2, entries 5–6). There is no significant increase in yield by further extension of the reaction time above one hour (Table 2, entries 7–8). By adjusting the ratio of the two starting materials, the highest yield, which was 95%, was obtained when the ratio of 1a/2a was 1.1/1 (Table 2, entries 9–11).
2.1.2. The Effect of Different Amide N-Leaving Groups on the Reaction
After establishing the optimal reaction conditions, we tested the effect of different amide N-leaving groups on the reaction, focusing primarily on the influence of various alkyl and aryl groups (R_1_ and R_2_) on the nitrogen atom in amides (Scheme 2). At first, we examined the impact of different functional groups (R_1_ and R_2_) on the nitrogen atom of benzylamides. Unfortunately, the reaction only worked best when the nitrogen atom contained the morpholine functional group (Scheme 2, 1a). In cases where other functional groups (R_1_ and R_2_) were present on the nitrogen, the yields ranged from 22% to 67% (Scheme 2, 1m–1q). In order to verify these findings, we obtained similar results by substituting the benzamides for benzylamides (Scheme 2, 1r–1t). This variation in yield is probably due to the polar oxygen atom in morpholine.
2.1.3. Substrates’ Scope Under Optimized Conditions
Under the optimal reaction conditions, we explored the scope of amide compounds in the reaction. As shown in Scheme 3, amides (1) with different alkyl groups (R_3_) have been reacted with 2-aminophenol (2a) to give the corresponding products (3). The yields ranged from moderate to excellent. When the benzyl group contains various functional groups, such as methyl, methoxy, or halogen groups, the reaction generally proceeds with excellent results (3aa–3ah). No significant steric or electronic effects were observed over the above substrates. It is noteworthy that other alkyl amides, such as isopropyl and cycloalkyl amides, also reacted efficiently (3ai–3aj). Excitingly, the alkylamides containing halogens and alkenes also led to the desired products (3ak–3al). This significantly extends the scope of 2-alkylbenzoxazole synthesis. The same method can also be used to prepare 2-arylbenzooxazole with a yield of 87% (3ar). This can be achieved by using benzamide as the starting material. To demonstrate the potential usefulness of the method, we carried out the reaction on a gram scale and obtained the product 3aa in a yield comparable to that of the small-scale reaction.
We also explored the effects of different substituents on the nucleophiles in the reaction (Scheme 4). Alkyl-substituted 2-aminophenols, such as methyl and tert-butyl 2-aminophenols, can be used to give good yields of 2-benzylbenzoxazoles (3ba–3da). If the 2-aminophenols contains halogen substituents, such as chlorine or bromine, the target product is also obtained in excellent yields (3ea–3fa). The nitro-substituted aminophenol compound shows an impressive tolerance in this reaction and the desired products are produced with an ideal yield of 65% (3ga). Despite a moderate yield, an aromatic pyridine substrate also gives the target product (3ha). In particular, replacing the aminophenol group with o-thiosamine leads to the formation of 2-alkylbenzothiazole (3ia). This greatly expands the synthetic scope of benzoheterocyclic compounds. We also tested several special substituents of 2-aminophenol, such as amino groups, sulfonamide groups, or ester groups. The experimental results found that under the action of Tf_2_O, the amino and amide groups form amidine, and the esters can consume anhydrides, which makes it difficult to continue the reaction. (3ja–3la).
2.2. Proposed Mechanism
Based on the experimental results and previous studies [26,45], we proposed a possible reaction mechanism for the synthesis of 2-substituted benzoxazole (Scheme 5). First of all, the amide (1) reacts with Tf_2_O and 2-Fluoropyridine to form an intermediate amidinium salt A. Then, the amino nitrogen of the 2-aminophenol compound (2a) acts as a nucleophile and attacks the carbon of the amidinium to form the intermediate C. In this step, one molecule of B is released by the addition/elimination reaction. Next, an intramolecular cyclization reaction is carried out to produce the intermediate D. Finally, the target product 3 is obtained by elimination reaction.
3. Materials and Methods
3.1. Materials and General Methods
The reaction vessel uses a thick-walled pressure bottle (upper pressure limit: 6 bar; reactor volume: 15 mL or 350 mL). All reagents used in experiment were obtained from commercial sources and used without further purification. Chromatographic purification of products was accomplished using silica gel (200–300 mesh). All melting points were determined on a Yanaco MP-500 micro melting point apparatus (Yanaco, Kyoto, Japan) and were uncorrected. All spectra of ^1^H NMR (400 MHz) and ^13^C NMR (100 MHz) were recorded on a JEOL JNM-ECA 400 spectrometer (JEOL, Tokyo, Japan) in CDCl_3_. NMR spectrum data processing using Delta NMR processing and control software (5.3.1 Windows). TMS was used as an internal reference and J values are given in Hz. PE is petroleum ether (60–90 °C).
3.2. General Procedure for the Preparation of 2-Substituted Benzoxazoles 3
2-Fluoropyridine (1 mmol, 97 mg) was added to a solution of amide 1a (0.55 mmol, 113 mg) in 1 mL DCM. The mixture was cooled to 0 °C and Tf_2_O (0.6 mmol, 170 mg) was added dropwise and stirred for 15 minutes. Then, 2-aminophenol 2a (0.5 mmol, 54.5 mg) was added and the reaction was stirred for 1 hour at room temperature. The reaction was quenched with 0.5 mL Et_3_N. The solvent was evaporated and the residue was purified by chromatography (silica gel, PE: EtOAc = 20:1) to give the desired product 2-benzylbenzo[d]oxazole (3aa, 99 mg, 95%) as a yellowish oil.
The products 3ab–3ar and 3ba–ia were prepared by similar procedure.
3.3. Analytical Data of Compound 3aa–3ar and 3ba–ia
2-Benzylbenzo[d]oxazole (3aa) [46]. The solvent was evaporated and the residue was purified by chromatography (silica gel, PE: EtOAc = 20:1) to give the desired product 3aa (99 mg, 95%) as a yellowish oil. ^1^H NMR (400 MHz, CDCl_3_) δ 7.72–7.64 (m, 1H), 7.45–7.40 (m, 1H), 7.39–7.28 (m, 4H), 7.28–7.21 (m, 3H), 4.24 (s, 2H). ^13^C NMR (100 MHz, CDCl_3_) δ 165.1, 150.9, 141.3, 134.7, 128.9 (2C), 128.7 (2C), 127.2, 124.6, 124.1, 119.7, 110.3, 35.2.
2-(4-Methylbenzyl)benzo[d]oxazole (3ab) [46]. The solvent was evaporated and the residue was purified by chromatography (silica gel, PE:EtOAc = 20:1) to give the desired product 3ab (100 mg, 90%) as a yellowish oil. ^1^H NMR (400 MHz, CDCl_3_) δ 7.70–7.65 (m, 1H), 7.47–7.41 (m, 1H), 7.31–7.23 (m, 4H), 7.17–7.11 (m, 2H), 4.22 (s, 2H), 2.32 (s, 3H). ^13^C NMR (100 MHz, CDCl_3_) δ 165.4, 151.0, 141.3, 136.9, 131.7, 129.5 (2C), 128.8 (2C), 124.6, 124.1, 119.8, 110.4, 34.8, 21.0.
2-(3-Methylbenzyl)benzo[d]oxazole (3ac) [46]. The solvent was evaporated and the residue was purified by chromatography (silica gel, PE:EtOAc = 20:1) to give the desired product 3ac (89 mg, 80%) as a yellowish oil. ^1^H NMR (400 MHz, CDCl_3_) δ 7.72–7.65 (m, 1H), 7.47–7.42 (m, 1H), 7.31–7.14 (m, 5H), 7.10–7.05 (m, 1H), 4.22 (s, 2H), 2.33 (s, 3H).^13^C NMR (100 MHz, CDCl_3_) δ 165.3, 151.0, 141.3, 138.5, 134.6, 129.7, 128.7, 128.0, 126.0, 124.6, 124.1, 119.8, 110.4, 35.2, 21.3.
2-(2-Methylbenzyl)benzo[d]oxazole (3ad) [46]. The solvent was evaporated and the residue was purified by chromatography (silica gel, PE:EtOAc = 20:1) to give the desired product 3ad (106 mg, 95%) as a yellow oil. ^1^H NMR (400 MHz, CDCl_3_) δ 7.70–7.64 (m, 1H), 7.45–7.39 (m, 1H), 7.31–7.22 (m, 3H), 7.22–7.14 (m, 3H), 4.24 (s, 2H), 2.38 (s, 3H). ^13^C NMR (100 MHz, CDCl_3_) δ 165.0, 150.9, 141.3, 136.7, 133.2, 130.5, 129.9, 127.5, 126.3, 124.5, 124.0, 119.7, 110.3, 33.0, 19.6.
2-(4-Methoxybenzyl)benzo[d]oxazole (3ae) [46]. The solvent was evaporated and the residue was purified by chromatography (silica gel, PE:EtOAc = 20:1) to give the desired product 3ae (111 mg, 93%) as a yellowish oil. ^1^H NMR (400 MHz, CDCl_3_) δ 7.70–7.65 (m, 1H), 7.47–7.42 (m, 1H), 7.32–7.23 (m, 4H), 6.91–6.84 (m, 2H), 4.20 (s, 2H), 3.77 (s, 3H). ^13^C NMR (100 MHz, CDCl_3_) δ 165.5, 158.8, 151.0, 141.3, 130.0 (2C), 126.7, 124.6, 124.1, 119.7, 114.2 (2C), 110.4, 55.2, 34.4.
2-(4-Fluorobenzyl)benzo[d]oxazole (3af) [47]. The solvent was evaporated and the residue was purified by chromatography (silica gel, PE:EtOAc = 20:1) to give the desired product 3af (99 mg, 87%) as a yellowish oil. ^1^H NMR (400 MHz, CDCl_3_) δ 7.71–7.65 (m, 1H), 7.48–7.42 (m, 1H), 7.37–7.24 (m, 4H), 7.06–6.98 (m, 2H), 4.23 (s, 2H). ^13^C NMR (100 MHz, CDCl_3_) δ 164.9, 162.1 (d, J = 244.1 Hz), 151.0, 141.3, 130.5 (2C, d, J = 7.6 Hz), 130.4 (d, J = 3.8 Hz), 124.8, 124.2, 119.8, 115.7 (2C, d, J = 21.9 Hz), 110.4, 34.4.
2-(4-Chlorobenzyl)benzo[d]oxazole (3ag) [25]. The solvent was evaporated and the residue was purified by chromatography (silica gel, PE:EtOAc = 20:1) to give the desired product 3ag (114 mg, 94%) as a white oil. ^1^H NMR (400 MHz, CDCl_3_) δ 7.71–7.65 (m, 1H), 7.48–7.42 (m, 1H), 7.33–7.24 (m, 6H), 4.22 (s, 2H). ^13^C NMR (100 MHz, CDCl_3_) δ 164.6, 151.0, 141.2, 133.3, 133.1, 130.3 (2C), 128.9 (2C), 124.8, 124.2, 119.8, 110.4, 34.5.
2-(4-Bromobenzyl)benzo[d]oxazole (3ah) [29]. The solvent was evaporated and the residue was purified by chromatography (silica gel, PE:EtOAc = 20:1) to give the desired product 3ah (133 mg, 93%) as a white solid, mp 95−98 °C. ^1^H NMR (400 MHz, CDCl_3_) δ 7.71–7.65 (m, 1H), 7.48–7.42 (m, 3H), 7.32–7.21 (m, 4H), 4.20 (s, 2H). ^13^C NMR (100 MHz, CDCl_3_) δ 164.4, 151.0, 141.2, 133.6, 131.9 (2C), 130.7 (2C), 124.8, 124.2, 121.3, 119.8, 110.4, 34.6.
2-Isopropylbenzo[d]oxazole (3ai) [48]. The solvent was evaporated and the residue was purified by chromatography (silica gel, PE:EtOAc = 30:1) to give the desired product 3ai (55 mg, 68%, stirred in DCE at 90 °C for 1 h) as a brown oil. ^1^H NMR (400 MHz, CDCl_3_) δ 7.71–7.66 (m, 1H), 7.51 7.45 (m, 1H), 7.32–7.25 (m, 2H), 3.25 (heptuple, J = 6.9 Hz, 1H), 1.46 (d, J = 6.9 Hz, 6H). ^13^C NMR (100 MHz, CDCl_3_) δ 171.3, 150.7, 141.2, 124.4, 124.0, 120.0, 110.2, 28.9, 20.3 (2C).
2-(2-Cyclohexylethyl)benzo[d]oxazole (3aj) [26]. The solvent was evaporated and the residue was purified by chromatography (silica gel, PE:EtOAc = 30:1) to give the desired product 3aj (93 mg, 81%, stirred in DCE at 90 °C for 1 h) as a brown oil. ^1^H NMR (400 MHz, CDCl_3_) δ 7.69–7.63 (m, 1H), 7.50–7.44 (m, 1H), 7.32–7.24 (m, 2H), 2.98–2.89 (m, 2H), 1.82–1.61 (m, 7H), 1.39–1.10 (m, 4H), 1.05–0.88 (m, 2H). ^13^C NMR (100 MHz, CDCl_3_) δ 167.6, 150.7, 141.4, 124.3, 123.9, 119.4, 110.2, 37.1, 34.1, 32.9 (2C), 26.5, 26.1(3C).
2-(5-Bromopentyl)benzo[d]oxazole (3ak) [49]. The solvent was evaporated and the residue was purified by chromatography (silica gel, PE:EtOAc = 30:1) to give the desired product 3ak (63 mg, 47%, stirred in DCE at 90 °C for 1 h) as a white solid, mp 81−83 °C. ^1^H NMR (400 MHz, CDCl_3_) δ 7.70–7.64 (m, 1H), 7.51–7.45 (m, 1H), 7.33–7.26 (m, 2H), 3.42 (t, J = 6.6 Hz, 2H), 2.95 (t, J = 7.6 Hz, 2H), 1.98–1.88 (m, 4H), 1.64–1.54 (m, 2H). ^13^C NMR (100 MHz, CDCl_3_) δ 166.7, 150.7, 141.3, 124. 5, 124.1, 119.5, 110.2, 33.3, 32.3, 28.4, 27.6, 25.8.
2-(But-3-en-1-yl)benzo[d]oxazole (3al) [50]. The solvent was evaporated and the residue was purified by chromatography (silica gel, PE:EtOAc = 30:1) to give the desired product 3al (36 mg, 42%, stirred in DCE at 90 °C for 1 h) as a colorless oil. ^1^H NMR (400 MHz, CDCl_3_) δ 7.70–7.64 (m, 1H), 7.51–7. 45 (m, 1H), 7.32–7.25 (m, 2H), 6.00–5.82 (m, 1H), 5.17–5.08 (m, 1H), 5.08–5.00 (m, 1H), 3.03 (t, J = 7.6 Hz, 2H), 2.71–2.59 (m, 2H). ^13^C NMR (100 MHz, CDCl_3_) δ 166.4, 150.8, 141.3, 136.2, 124.4, 124.0, 119.5, 116.0, 110.2, 30.5, 28.1.
2-Phenylbenzo[d]oxazole (3ar) [26]. The solvent was evaporated and the residue was purified by chromatography (silica gel, PE:EtOAc = 15:1) to give the desired product 3ar (85 mg, 87%) as a white solid, mp 95−97 °C (lit. 97–98 °C). ^1^H NMR (400 MHz, CDCl_3_) δ 8.27–8.24 (m, 2H), 7.79–7.76 (m, 1H), 7.59–7.49 (m, 4H), 7.37–7.31 (m, 2H). ^13^C NMR (100 MHz, CDCl_3_) δ 162.9, 150.7, 142.1, 131.5, 128.9 (2C), 127.6 (2C), 127.1, 125.1, 124.5, 119.9, 110.5.
2-Benzyl-4-methylbenzo[d]oxazole (3ba) [33]. The solvent was evaporated and the residue was purified by chromatography (silica gel, PE:EtOAc = 20:1) to give the desired product 3ba (104 mg, 93%) as a colorless oil. ^1^H NMR (400 MHz, CDCl_3_) δ 7.41–7.29 (m, 4H), 7.29–7.22 (m, 2H), 7.20–7.12 (m, 1H), 7.11–7.05 (m, 1H), 4.27 (s, 2H), 2.61 (s, 3H).^13^C NMR (100 MHz, CDCl_3_) δ 164.2, 150.8, 140.5, 135.0, 130.1, 128.9 (2C), 128.7 (2C), 127.2, 124.7, 124.3, 107.7, 35.3, 16.5.
2-Benzyl-5-methylbenzo[d]oxazole (3ca) [33]. The solvent was evaporated and the residue was purified by chromatography (silica gel, PE:EtOAc = 20:1) to give the desired product 3ca (99 mg, 89%) as a yellowish oil. ^1^H NMR (400 MHz, CDCl_3_) δ 7.46 (s, 1H), 7.39–7.29 (m, 5H), 7.29–7.23 (m, 1H), 7.10–7.05 (m, 1H), 4.24 (s, 2H), 2.43 (s, 3H). ^13^C NMR (100 MHz, CDCl_3_) δ 165.2, 149.3, 141.5, 134.9, 133.9, 128.9 (2C), 128.7 (2C), 127.2, 125.7, 119.7, 109.8, 35.3, 21.4.
2-Benzyl-5-(tert-butyl)benzo[d]oxazole (3da) [46]. The solvent was evaporated and the residue was purified by chromatography (silica gel, PE:EtOAc = 20:1) to give the desired product 3da (115 mg, 87%) as a yellowish oil. ^1^H NMR (400 MHz, CDCl_3_) δ 7.72–7.69 (m, 1H), 7.40–7.29 (m, 6H), 7.29–7.22 (m, 1H), 4.24 (s, 2H), 1.36 (s, 9H). ^13^C NMR (100 MHz, CDCl_3_) δ 165.3, 149.0, 147.7, 141.2, 134.9, 128.9 (2C), 128.8 (2C), 127.2, 122.3, 116.3, 109.5, 35.3, 34.8, 31.7 (3C).
2-Benzyl-5-chlorobenzo[d]oxazole (3ea) [25]. The solvent was evaporated and the residue was purified by chromatography (silica gel, PE:EtOAc = 20:1) to give the desired product 3ea (116 mg, 95%) as a yellowish oil. ^1^H NMR (400 MHz, CDCl_3_) δ 7.66–7.65 (m, 1H), 7.39–7.31 (m, 5H), 7.31–7.23 (m, 2H), 4.25 (s, 2H). ^13^C NMR (100 MHz, CDCl_3_) δ 166.6, 149.6, 142.4, 134.3, 129.6, 129.0 (2C), 128.9 (2C), 127.4, 125.0, 119.8, 111.1, 35.2.
2-Benzyl-5-bromobenzo[d]oxazole (3fa) [33]. The solvent was evaporated and the residue was purified by chromatography (silica gel, PE:EtOAc = 20:1) to give the desired product 3fa (137 mg, 95%) as a yellowish oil. ^1^H NMR (400 MHz, CDCl_3_) δ 7.82–7.80 (m, 1H), 7.42–7.23 (m, 7H), 4.25 (s, 2H). ^13^C NMR (100 MHz, CDCl_3_) δ 166.4, 150.0, 142.9, 134.3, 128.9 (2C), 128.8 (2C), 127.7, 127.4, 122.8, 116.9, 111.7, 35.2.
2-Benzyl-5-nitrobenzo[d]oxazole (3ga) [51]. The solvent was evaporated and the residue was purified by chromatography (silica gel, PE:EtOAc = 15:1) to give the desired product 3ga (83 mg, 65%) as a yellow oil. ^1^H NMR (400 MHz, CDCl_3_) δ 8.58–8.56 (m, 1H), 8.29–8.25 (m, 1H), 7.57–7.55 (m, 1H), 7.42–7.27 (m, 5H), 4.32 (s, 2H). ^13^C NMR (100 MHz, CDCl_3_) δ 168.4, 154.5, 145.1, 141.7, 133.7, 129.0 (2C), 129.0 (2C), 127.7, 120.9, 116.2, 110.6, 35.2.
2-Benzyloxazolo[4,5-b]pyridine (3ha) [52]. The solvent was evaporated and the residue was purified by chromatography (silica gel, PE:EtOAc = 8:1) to give the desired product 3ha (73 mg, 69%) as a colorless solid, mp 93−95 °C (lit. 95–97 °C). ^1^H NMR (400 MHz, CDCl_3_) δ 8.55–8.50 (m, 1H), 7.77–7.69 (m, 1H), 7.43–7.38 (m, 2H), 7.38–7.32 (m, 2H), 7.31–7.26 (m, 1H), 7.26–7.20 (m, 1H), 4.33 (s, 2H). ^13^C NMR (100 MHz, CDCl_3_) δ 168.2, 155.7, 146.2, 143.1, 134.0, 129.0 (2C), 128.8 (2C), 127.4, 119.8, 118.0, 35.5.
2-Benzylbenzo[d]thiazole (3ia) [53]. The solvent was evaporated and the residue was purified by chromatography (silica gel, PE:EtOAc = 30:1) to give the desired product 3ia (72 mg, 64%) as a yellowish oil. ^1^H NMR (400 MHz, CDCl_3_) δ 7.99 (d, J = 8.0 Hz, 1H), 7.77 (d, J = 8.4 Hz, 1H), 7.47–7.40 (m, 1H), 7.38–7.25 (m, 6H), 4.43 (s, 2H). ^13^C NMR (100 MHz, CDCl_3_) δ 171.1, 153.2, 137.1, 135.6, 129.1 (2C), 128.8 (2C), 127.3, 125.9, 124.8, 122.7, 121.5, 40.6.
4. Conclusions
In conclusion, we have developed a new method for the synthesis of 2-substituted benzoxazoles from tertiary amides and 2-aminophenols in the presence of Tf_2_O and 2-Fluoropyridine. The 2-substituted benzoxazoles have been prepared by cascade reactions, including Tf_2_O-activated amides, nucleophilic addition, intramolecular cyclization, and elimination. The method was simple, mild, and effective for the synthesis of 2-substituted benzoxazoles and can be extended to the synthesis of 2-substituted benzothiazoles. Given its versatility and ease of use, we anticipate that this method will have broad applications in organic synthesis.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Singh S. Veeraswamy G. Bhattarai D. Goo J.-I. Lee K. Choi Y. Recent Advances in the Development of Pharmacologically Active Compounds that Contain a Benzoxazole Scaffold Asian J. Org. Chem.201541338136110.1002/ajoc.201500235 · doi ↗
- 2Arulmurugan S. Kavitha H.P. Vennila J.P. Review on the Synthetic Methods of Biologically Potent Benzoxazole Derivatives MINI-Rev. Org. Chem.20211876978510.2174/1570193 X 17999201020231359 · doi ↗
- 3Wong X.K. Yeong K.Y. A Patent Review on the Current Developments of Benzoxazoles in Drug Discovery Chem Med Chem 2021163237326210.1002/cmdc.20210037034289258 · doi ↗ · pubmed ↗
- 4Di Martino S. De Rosa M. The Benzoxazole Heterocycle: A Comprehensive Review of the Most Recent Medicinal Chemistry Developments of Antiproliferative, Brain-Penetrant, and Anti-inflammatory Agents Top. Curr. Chem.20243823310.1007/s 41061-024-00477-639432195 · doi ↗ · pubmed ↗
- 5Arakawa K. Inamasu M. Matsumoto M. Okumura K. Yasuda K. Akatsuka H. Kawanami S. Watanabe A. Homma K. Saiga Y. Novel Benzoxazole 2,4-Thiazolidinediones as Potent Hypoglycemic Agents. Synthesis and Structure-Activity Relationships Chem. Pharm. Bull.1997451984199310.1248/cpb.45.19849433768 · doi ↗ · pubmed ↗
- 6Tekiner-Gulbas B. Temiz-Arpaci O. Yildiz I. Altanlar N. Synthesis and in Vitro Antimicrobial Activity of New 2-[p-substituted-benzyl]-5-[substituted-carbonylamino]benzoxazoles Eur. J. Med. Chem.2007421293129910.1016/j.ejmech.2007.01.02217337097 · doi ↗ · pubmed ↗
- 7Oksuzoglu E. Temiz-Arpaci O. Tekiner-Gulbas B. Eroglu H. Sen G. Alper S. Yildiz I. Diril N. Aki-Sener E. Yalcin I. A Study on the Genotoxic Activities of Some New Benzoxazoles Med. Chem. Res.20081611410.1007/s 00044-007-9005-z · doi ↗
- 8Jiang J. Tang X. Dou W. Zhang H. Liu W. Wang C. Zheng J. Synthesis and Characterization of the Ligand Based on Benzoxazole and its Transition Metal Complexes: DNA-binding and Antitumor Activity J. Inorg. Biochem.201010458359110.1016/j.jinorgbio.2010.01.01120202687 · doi ↗ · pubmed ↗