Synthesis of 2-Amino-4, 5-Diarylthiazole Derivatives and Evaluation of Their Anti-Candida Albicans Activity
Dongmei Gao, Lele Shi, Yuhang Huang, Yingmei Lv, Xuan Yang, Zhenting Du

TL;DR
This paper reports the synthesis of new thiazole compounds with antifungal activity against Candida albicans.
Contribution
The study introduces a new class of 2-amino-4,5-diarylthiazole derivatives with promising anti-Candida activity and provides insights into their mechanism of action.
Findings
Four derivatives showed moderate anti-Candida albicans activity in initial tests.
Compound 5a8 exhibited antifungal activity comparable to fluconazole (MIC80 = 9 μM).
Molecular docking studies suggested interactions with key antifungal target proteins.
Abstract
The thiazole heterocycle is one of the most common moieties found in various drugs. Using 2-aminothiazole as the core structure, the amino group was functionalized with an amide. As a result, 30 trisubstituted 2-amino-4, 5-diarylthiazole derivatives were synthesized, with different substitutions introduced at the C2, C4, and C5 positions. The anti-Candida albicans biological activities of these synthetic compounds on five kinds of Candida albicans at different concentrations were detected by the microdilution method. In the first round, four derivatives of 2-amino-4, 5-diarylthiazole exhibited moderate anti-Candida albicans activity. Among them, 4a8 was chosen to be subjected to a demethylation process. Thus, 5a8 was synthesized successfully, giving anti-Candida albicans activity (MIC80 = 9 μM) similar to that of a typical antifungal drug, fluconazole. To understand the mechanism of…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
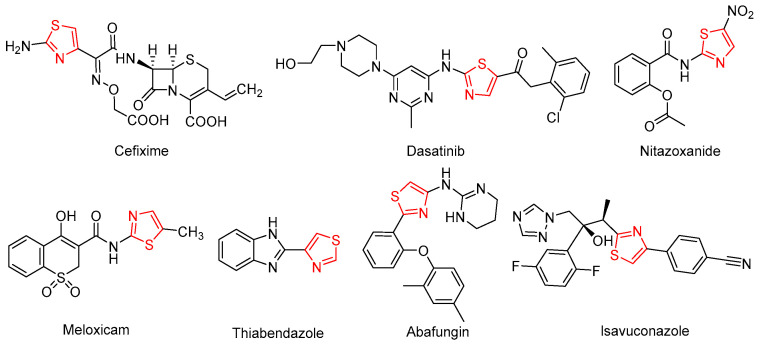 Figure 1
Figure 1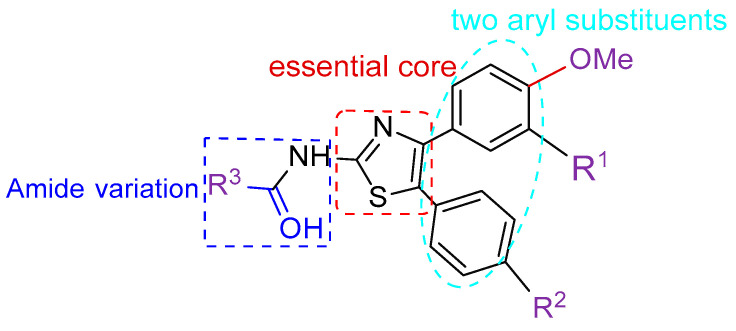 Figure 2
Figure 2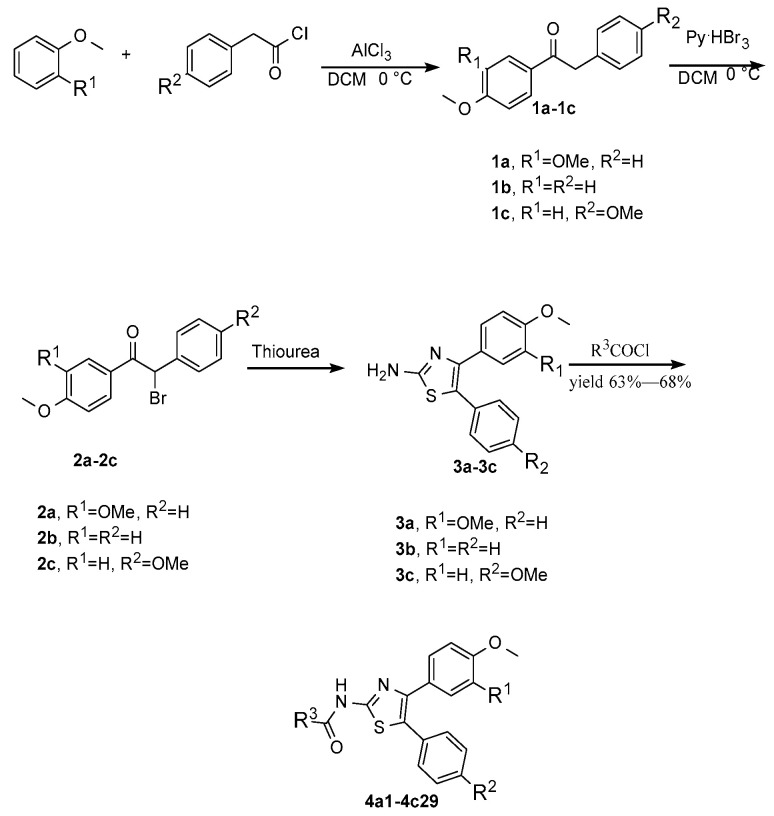 Figure 3
Figure 3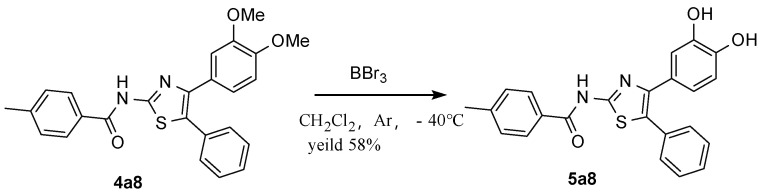 Figure 4
Figure 4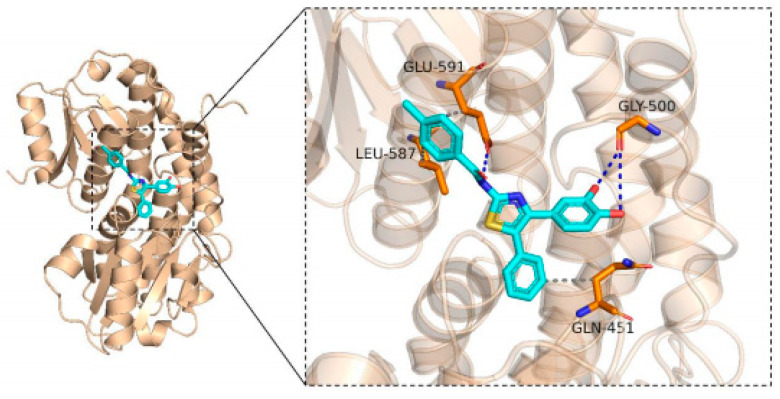 Figure 5
Figure 5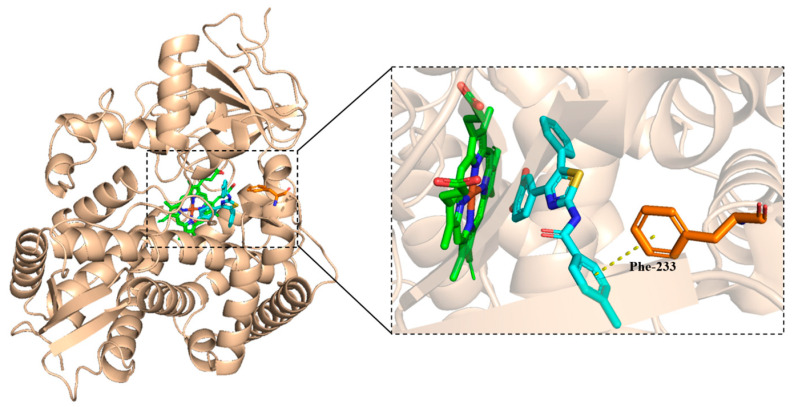 Figure 6
Figure 6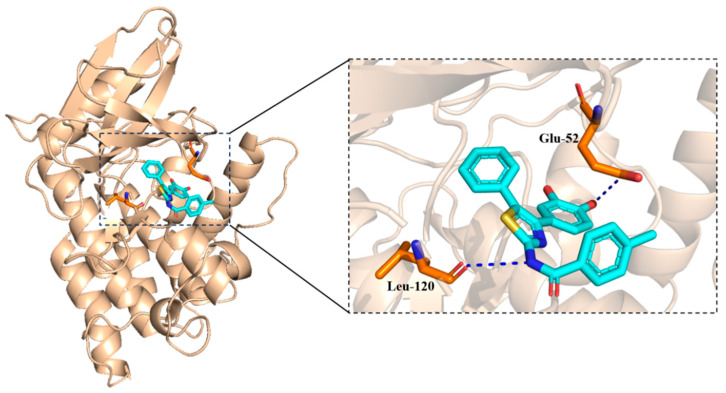 Figure 7
Figure 7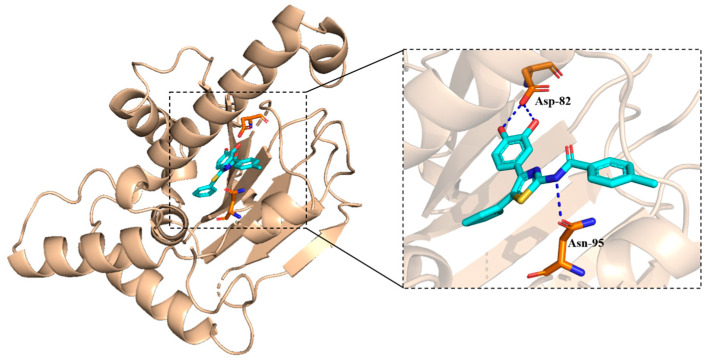 Figure 8
Figure 8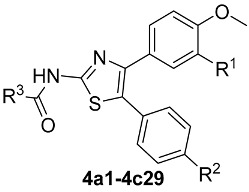 Figure 9
Figure 9|
| |
- —Science and Technology Program Projects of Yangling Demonstration Zone
- —Yangling Vocational & Technical College
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAntifungal resistance and susceptibility · Synthesis and Biological Evaluation · Synthesis and biological activity
1. Introduction
The incidence of invasive fungal infections (IFIs) has risen significantly, posing a serious clinical threat, largely due to the growing number of immunocompromised individuals worldwide [1,2,3]. While various factors contribute to this increase, a primary cause is the alteration of hosts’ immune systems, often a result of receiving solid organ or hematopoietic stem cell transplants, or the use of immune-modulating treatments such as tumor necrosis factor antagonists to manage chronic inflammatory conditions [4]. Among these infections, Candidiasis has emerged as one of the most frequent fungal diseases observed in immunosuppressed patients over the past two decades [5,6,7]. Such individuals are highly vulnerable to opportunistic infections, which are influenced by their specific health statuses. Infections caused by Candida albicans are particularly common in immunocompromised populations, such as those who have undergone long-term or heavy usages of broad-spectrum antibiotics, hormones, or immunosuppressive agents [8], or those with severe underlying conditions or history of organ transplant surgeries [9]. Currently, antifungal medications are the main therapeutic approach for deep fungal infections, but two significant challenges persist. First, prolonged use of antifungal drugs often leads to the development of drug resistance [10,11,12]. Second, the widespread emergence of drug-resistant bacteria in clinical settings has rendered antibiotic resistance a growing public health concern [13]. This has heightened the urgency for developing new antifungal agents or innovative drug delivery systems to address deep fungal infections [14,15,16].
Thiazole heterocycles represent a common structural motif in various biologically active compounds [17]. Numerous commercial drugs featuring the thiazole group (Figure 1), such as cefixime [18], dasatinib [19], nitazoxanide [20], meloxicam [21], abafungin [22] and isavuconazole [23], are widely employed in clinical antifungal treatments [24]. In the agricultural sector, thiabendazole [25,26] has been used as an antifungal agent against plant pathogens since Merck patented it in 1962. Bikobo and colleagues designed and synthesized a series of 2-phenylaminothiazole derivatives, evaluating their antifungal activity against Candida albicans and Candida krusei [27], and identified a promising lead compound with superior efficacy compared to fluconazole. Lino et al. synthesized novel thiazole derivatives with hydrazone groups and assessed their activity against seven fungi, including Candida albicans, with results indicating that the thiazole ring is crucial for antifungal effectiveness [28]. Kamat et al. developed pyridine-thiazole hydrazide compounds and evaluated their antibacterial and anti-inflammatory properties, finding that three of these compounds exhibited antifungal activity comparable to fluconazole and nystatin against four fungi, including Candida albicans [29].
Arora et al. obtained novel 2,4-disubstituted thiazole derivatives with anti-Candida albicans activity through computer-aided drug design (CADD) using the GFAT protein as a template, which laid a theoretical foundation for the study of the antifungal function of thiazole derivatives [30]. Pricopie et al. designed and synthesized two thiazole derivatives with lipophilic substituents and evaluated their anti-Candida activity in vitro. They found that the newly synthesized compounds may interfere with the ergosterol biosynthesis pathway by inhibiting 14α-demethylase (CYP51), and the destruction of the fungal cell membrane was observed through staining [31]. Cowen’s group screened a library of 736 protein kinase inhibitors, and they found that the 2,3-arylpyrazolopyridine derivative GW461484A was capable of restoring caspofungin sensitivity and potentiating the activity of fluconazole against Candida albicans. Yck2, a member of the CK1 (casein kinase 1) family, was confirmed to be the molecular target of GW461484A [32]. Based on the aforementioned literature review, thiazole derivatives exhibit notable antifungal activity, with 2,4-disubstituted aminothiazole compounds demonstrating significant antibacterial activity against clinical fungi Candida albicans. However, to the best of our knowledge, trisubstituted thiazole compounds are seldom reported. Heat-shock protein 90 (Hsp90) is an essential molecular chaperone that is highly conserved among eukaryotes [33]. Based on these results, studying how medicine candidates interact with these proteins is quite reasonable. Obviously, it was proved that using these targets to explore new leading compounds saves labor.
Thus, we envisioned that the 2-aminothiazole moiety would be a promising substructure as developing new anti-Candida albicans derivatives. In addition, introducing an amide bond to 2-aminothiazole is a common strategy in structural modifications. A series of different substituted benzamide, heterocyclic amide, and aliphatic amide are introduced at the C2 position, while two substituted aryl groups are incorporated at the C4 and C5 positions (Figure 2).
2. Results and Discussion
2.1. Synthetic Results
The synthetic workflow is shown in Figure 3. The synthesis commenced at the two-aryl moiety through a Friedel–Crafts reaction to 1,2-diaryl-ethanone, similar to what has been reported in the literature [34]. At 0 °C, they were subjected to a bromination process using pyridinium tribromide, which is an alternative to liquid bromine; the corresponding bromo-ethanone derivatives 2a–2c could be obtained in excellent yields. In ethanol, these derivatives reacted with thiourea at refluxed temperature, and the 2-NH_2_-thiazole heterocycles 3a–3c were constructed. None of these synthetic intermediates were characterized fully, and after a conventional amide formation using acyl chloride in the presence of triethyl amine, the final derivatives were catered in good overall yields. A total of 29 compounds were synthesized, as shown in Table 1.
2.2. Antifungal Activity Against Candida Albicans
After synthesizing 29 compounds, the antifungal activities of the designed derivatives against five species of Candida albicans were evaluated in vitro. We used an FLC-sensitive strain (Candida albicans 186382) and five FLC-resistant species (4935, 5122, 5172, and 5272) to evaluate the antifungal activity of our synthetic compounds alone according to microdilution methods. Interestingly, compounds 4a1, 4a8, 4b19, and 4b23 exhibited moderate activity (Table 2).
With these data on hand, to differentiate the activities between -OMe and -OH, the demethylated form of the most active 4a8 was obtained after it was subjected to a boron tribromide demethylation protocol [35] (Figure 4).
As shown in Table 3, after the demethylation, the corresponding compound showed excellent biological activity against Candida albicans. Regardless of whether the strain was sensitive or resistant, the activity was boosted remarkably in both. Even more, in this experimental condition, the biological activity was similar to that of the control, commercial fluconazole. It is possible that the two hydroxyl groups in 5a8 enhanced the solubility of the molecules and gave the best results. At the same time, the C2 position of the thiazole, aryl, especially 4-methyl phenyl, as in 5a8, had the best activity.
2.3. Molecular Docking Against Different Proteins
The molecular docking method was used to predict the binding mode between the small molecule and the protein. The docking process was carried out using AutoDock Vina software(v1.1.2). Before docking, the crystal structure of Candida albicans glutamine-fructose-6-phosphate aminotransferase (GFAT), protein kinase (Yck2), lanosterol 14a-demethylase (CYP51), and heat-shock protein 90 (Hsp90) were obtained from the Protein Data Bank (PDB) (PDB ID, 2POC). The 3D structure of compound 5a8 was constructed using Chem3D, and energy-minimized with the MMFF94 force field. The protein was preprocessed using PyMol(V2.4.0a0) software, including hydrogen addition, the removal of water molecules, and the removal of non-ligand small molecules. A docking box was then defined to enclose compound 5a8 within the protein’s active pocket. Finally, the PDB format of both the small molecule and receptor protein was converted to PDBQT format using the AutoDockFR software(v1.0) suite. The docking was then performed, and the results were visualized and analyzed using PyMol.
2.3.1. GFAT as the Target
Glutamine-fructose-6-phosphoamidamitransferase (GFAT) was found to be the key rate-limiting enzyme in the first step of chitin synthesis in the cell walls of fungi [30]. Inhibition of GAFT leads to the inability of the fungal cell wall to grow, resulting in the death of the entire fungus.
In Figure 5, the blue color represents the 3D structure of small molecule 5a8, the orange color represents the protein GFAT structure, the blue dashed line represents hydrogen bonding, and the gray dashed line represents the hydrophobic interaction. Thus, we could see that compound 5a8 is embedded into the protein GFAT. It interacts with the residual of Gly-500 and Glu-591 by hydrogen bond. It also maintains hydrophobic interaction with Leu-587, Gln-451, and Gln-591. This reveals that the hydroxyl group is indispensable in the exhibition of biological activities. The docking score was −7.426 kcal/mol.
2.3.2. CYP51 as Target
There is some π-π stack between the methylphenyl group and Phe-233 in CYP51. However, the docking score was as high as −8.104 kcal/mol, as shown in Figure 6.
2.3.3. Yck2 as Target
As shown in Figure 7, two hydrogen bonds (blue dash line) were observed. The hydroxyl group seemed important in forming the hydrogen bonds. The docking score was −4.894 kcal/mol.
2.3.4. Hsp90 as Target
As shown in Figure 8, there were three main interactions between 5a8 and Hsp90. These interactions revealed that the hydroxyl group in 5a8 is crucial for biological activity. The docking score was −6.431 kcal/mol.
3. Materials and Methods
Melting points were determined using a WRS-1B melting point apparatus (Shanghai Precision & Scientific Instrument Co., Ltd., Shanghai, China); HRMS data were acquired on a Waters Xevo G2-S QTof mass spectrometer (Waters Corporation, Milford, MA, USA) with an electrospray ionization (ESI) source; and NMR spectra were recorded on Bruker Avance III 400 and 500 MHz spectrometers (Bruker Biospin, Rheinstetten, Germany) equipped with 5 mm inverse probes. The general procedures are as follows: Air- and moisture-sensitive reactions were performed in flame-dried round bottom flasks fitted with rubber septa under a positive argon pressure. Air- and moisture-sensitive liquids and solutions were transferred via a syringe or stainless-steel cannula. Organic solutions were concentrated by rotary evaporation below 35 °C at 20 mm Hg. Flash column chromatography was performed employing silica gel (230–400 mesh). Thin-layer chromatography (TLC) was performed using glass plates with silica gel F245, and eluted by PE:EA 20:1–5:1. HRMS was detected on a Waters G2-S qtof instrument. All chemicals were purchased from Chinese Chemical Company such as Aladdin, Leyan, Energy chemiccls etc.
3.1. 1-(3,4-Dimethoxyphenyl)-2-phenylethan-1-one 1a
To a 250 mL round-bottom flask, o-dimethoxybenzene (10.0 g, 72.38 mmol) in 100 mL of dichloromethane was added. Then, anhydrous aluminum chloride (14.5 g, 108.57 mmol) was added to the flask quickly; phenylacetyl chloride (12.5 mL, 94.09 mmol) was introduced into the reaction flask dropwise. The reaction was allowed to stir at 0 °C for two hours and was monitored by TLC. After the reaction was complete, it was quenched by pouring into ice water. After the removal of the reaction solvent under reduced pressure, the residual was extracted with ethyl acetate (100 mL × 3). The extracts were combined, dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. Compound 1a was obtained in 78% yield after column chromatography.
The synthesis of 1b and 1c was similar to that of 1a, and in the yields of 74% and 76%, respectively.
3.2. 2-Bromo-1-(3,4-dimethoxyphenyl)-2-phenylethan-1-one 2a
At first, the reaction glassware was equipped with an alkalic absorption beaker. To that, under an ice-water bath, a solution of 1a (14.0 g, 54.62 mmol) in dichloromethane (100 mL) was added to pyridinium tribromide (19.0 g, 59.41 mmol), and the reaction mixture was allowed to stir for five hours at ambient temperature. The reaction process was monitored by TLC. After completion, it was quenched with sodium sulfite; the reaction mixture was extracted with ethyl acetate (100 mL × 3). The extracts were combined, dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. Compound 2a was used without further purification.
The synthesis of 2b and 2c was similar to that of 1a, with yields of 72% and 70%, respectively.
3.3. 4-(3,4-Dimethoxyphenyl)-5-phenylthiazol-2-amine 3a
To a 250 mL round-bottom flask, 2a (15.0 g, 49.48 mmol) and 95% ethanol (100 mL) were added, before adding thiourea (4.1 g, 54.43 mmol), and the reaction mixture was heated to reflux for five hours. The reaction was monitored by TLC. After the reaction was complete, it was quenched by pouring into ice water. After the removal of the most reaction solvent under reduced pressure, the residual was basified to pH 7–8 using 10% NaOH, and then a large quantity of solid precipitated. It was filtered, collected, and dried. Compound 3a was obtained in 70% yield after column chromatography using PE:EA 20:1–5:1 as eluents.
The synthesis of 3b and 3c was similar to that of 1a, with yields of 72% and 70%, respectively.
3.4. N-(4-(3,4-dimethoxyphenyl)-5-phenylthiazol-2-yl)butyramide 4a1
To a solution of intermediate 3a (0.3 g, 0.96 mmol) in 20 mL tetrahydrofuran, an appropriate amount of triethylamine and a small amount of 4-dimethylamino pyridine was added as catalysts. Finally, butyryl chloride (0.14 mL, 1.15 mmol) was introduced into the above-mentioned mixed system, and the reaction was allowed to stir at room temperature for about 10 h. The reaction was monitored by TLC until completion. The reaction mixture was poured into ice water, and the precipitate was collected and recrystallized in methanol. The target compound 4a1 was obtained with a 65% yield.
The synthesis of compounds 4a2–4c29 was carried out using a procedure analogous to that described for 4a1, yielding target products in 63–68% isolated yields. Comprehensive structural characterization data for all new compounds—including ^1^H NMR, ^13^C NMR, and HRMS spectra—are provided in the Supplementary Information.
3.5. N-(4-(3,4-dihydroxyphenyl)-5-phenylthiazol-2-yl)-4-methylbenzamide 5a8
To a solution of 4a8 (0.3 g, 0.72 mmol) reacting with 20 mL of dichloromethane in a 50 mL round-bottom flask, at −56 °C, boron tribromide (0.35 mL, 3.60 mmol) was added by syringe. The reaction was monitored by TLC. After completion, the reaction was quenched by a small amount of methanol. After removal of the dichloromethane under reduced pressure, the residue was washed by water and brine, dried over anhydrous Na_2_SO_4_, and filtered. The filtrate was evaporated under vacuum to obtain compound 5a8 (yield 72%) as yellowish solid, m. p. 123.6–125.5 °C.
3.6. Antifungal Activity In Vitro
Determination of MIC80 and MFC Values
The antifungal activities in vitro of target compounds against five strains of fungi were determined via the BMD method with assays in 96-well plates as described in the CLSI guidelines (CLSI M27-A3 and M38-A2) [36,37,38]. FLC was selected as the positive control. To prepare the stock solutions of the tested compounds, they were dissolved in 100% DMSO with a concentration of 0.8 mg/mL. Before the assay, the solutions were diluted to a concentration of 8.0 µg/mL with RPMI medium 1640. The final concentrations of tested compounds were 64, 32, 16, 8, 4, 2, 1, 0.5, 0.25, and 0.125 µg/mL. After inoculation, the plates were incubated at 30 °C for 24 h. The absorbance of each well was scanned at a wavelength of 625 nm via an ELISA reader (DNM 9602, Perlong, Bingjing, China), and the inhibition rate was calculated.
where AP is the absorbance of positive control wells, AD is the absorbance of drug wells, and AN is the absorbance of negative control wells. The MIC_80_ values were the concentration of tested compounds when the inhibition rate was at 80%. A curve was plotted with the logarithm of the concentration as the horizontal axis and the inhibition rate as the vertical axis, and the MIC_80_ was calculated.
Among them, A refers to the corresponding minimum concentration in the aforementioned gradient concentrations when the inhibition rate is just above 80%. a is the inhibition rate at concentration A, b is the inhibition rate just below a, and N represents the dilution factor.
4. Conclusions
Thirty novel trisubstituted 2-aminothiazole derivatives were efficiently synthesized by introducing different substituents (including benzamides, heterocyclic amides, and fatty amides) at the C2 position of the thiazole ring. Substituted aryl groups were also incorporated at the C4 and C5 positions with 2-aminothiazole as the parent nucleus. In the first round of experiments, the promising compound 4a8 was found to exhibit the best activity among the 29 candidates. Consequently, its demethylated derivative (compound 5a8) was synthesized, and demonstrated activity comparable to fluconazole in anti-Candida albicans assays. Molecular docking calculations targeting four proteins were performed to gain insight into the anti-Candida albicans activity of 5a8. The biological activity is primarily attributed to π-π stacking, hydrophobic interactions, and hydrogen bonding.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Brown G.D. Denning D.W. Gow N.A.R. Levitz S.M. Netea M.G. White T.C. Hidden Killers: Human Fungal Infections Sci. Transl. Med.20124 rv 113rv 16510.1126/scitranslmed.300440423253612 · doi ↗ · pubmed ↗
- 2Brown G.D. Denning D.W. Levitz S.M. Tackling Human Fungal Infections Science 201233664710.1126/science.122223622582229 · doi ↗ · pubmed ↗
- 3Perfect J.R. The antifungal pipeline: A reality check Nat. Rev. Drug Discov.20171660361610.1038/nrd.2017.4628496146 PMC 5760994 · doi ↗ · pubmed ↗
- 4Lockhart S.R. Guarner J. Emerging and reemerging fungal infections Semin. Diagn. Pathol.20193617718110.1053/j.semdp.2019.04.01031010605 PMC 11979780 · doi ↗ · pubmed ↗
- 5Yapar N. Epidemiology and risk factors for invasive Candidiasis Ther. Clin. Risk Manag.2014109510510.2147/TCRM.S 4016024611015 PMC 3928396 · doi ↗ · pubmed ↗
- 6Spampinato C. Leonardi D. Candida Infections, Causes, Targets, and Resistance Mechanisms: Traditional and Alternative Antifungal Agents Bio Med Res. Int.2013201320423710.1155/2013/20423723878798 PMC 3708393 · doi ↗ · pubmed ↗
- 7Nguyen W. Grigori L. Just E. Santos C. Seleem D. The in vivo anti-Candida albicans activity of flavonoids J. Oral Biosci.20216312012810.1016/j.job.2021.03.00433839266 · doi ↗ · pubmed ↗
- 8Drummond R.A. Desai J.V. Ricotta E.E. Swamydas M. Deming C. Conlan S. Quinones M. Matei-Rascu V. Sherif L. Lecky D. Long-term antibiotic exposure promotes mortality after systemic fungal infection by driving lymphocyte dysfunction and systemic escape of commensal bacteria Cell Host Microbe 20223010201033.e 102610.1016/j.chom.2022.04.01335568028 PMC 9283303 · doi ↗ · pubmed ↗