Synthesis and Antimicrobial Activity of Canthin-6-One Alkaloids
Xubing Qi, Yogini Jaiswal, Xinrong Xie, Yu Fan, Rongping Wu, Shaoyang Su, Yifu Guan, Leonard Williams, Xun Song

TL;DR
Scientists synthesized new canthin-6-one alkaloids and tested their antimicrobial effects against several pathogens.
Contribution
The study reports the first synthesis of four canthin-6-one alkaloids and eight new derivatives, along with their antimicrobial activity against human pathogens.
Findings
Seventeen compounds, including nine canthin-6-ones, were synthesized and structurally confirmed.
Alkaloids 1-4 and 9 showed antimicrobial activity against C. albicans, C. neoformans, and S. aureus at 50 µg/mL.
The antimicrobial activity of compounds 2, 4-5, and 12-13 against four human pathogens is reported for the first time.
Abstract
Canthin-6-one alkaloids have consistently attracted the interest of medicinal chemists due to their wide range of promising bioactivities, including antitumor, antifungal, antibacterial, and antiviral properties. However, their low natural abundance in plants has constrained the further exploration of their potential bioactivities. This study reports a comprehensive synthesis of canthin-6-one alkaloids, utilizing key Suzuki coupling and Cu-catalyzed amidation reactions to construct their core scaffold. Derivatives were synthesized with Koenig–Knorr glycosylation for the further modification of synthetic canthin-6-ones. The antimicrobial activities of the synthesized compounds were evaluated against C. albicans, C. neoformans, S. aureus and E. coli using the micro-dilution method. In total, 17 compounds were synthesized, including nine canthin-6-ones. Notably, alkaloids 4, 5, 7 and 12-13…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
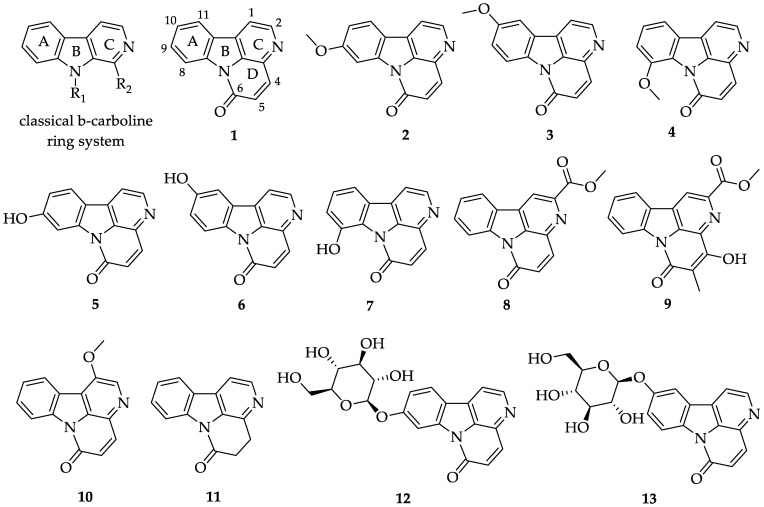 Figure 1
Figure 1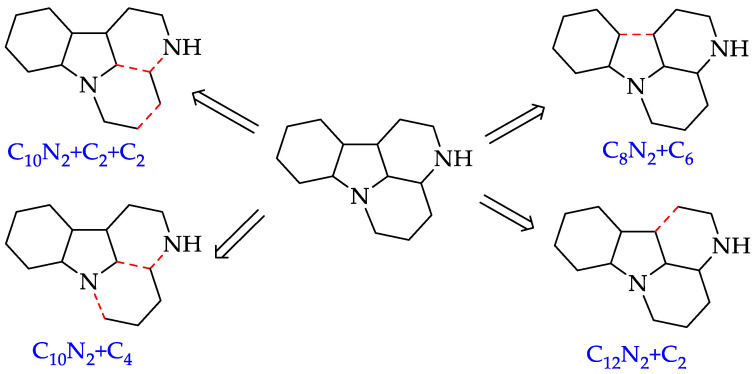 Figure 2
Figure 2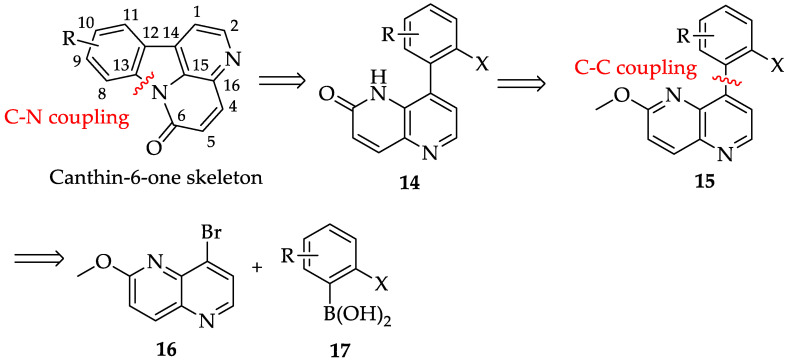 Figure 3
Figure 3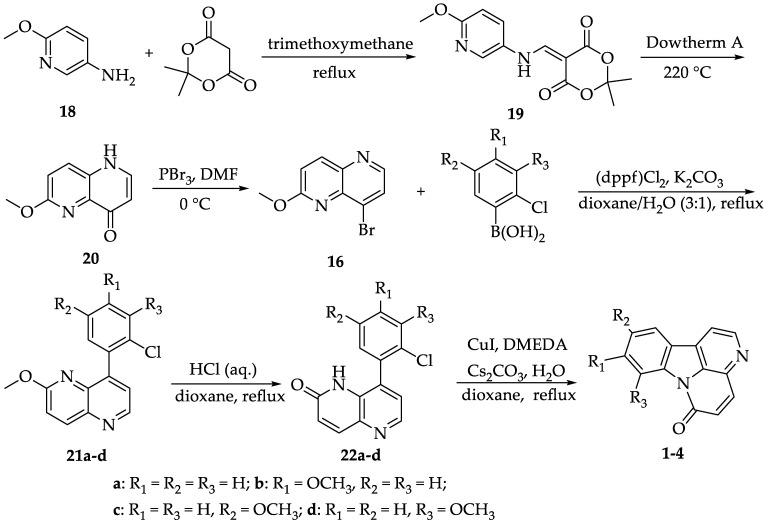 Figure 4
Figure 4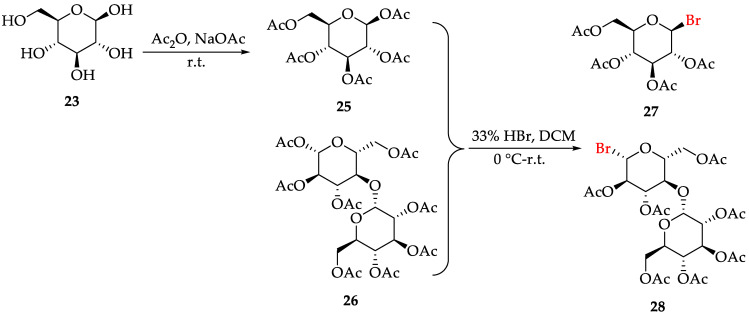 Figure 5
Figure 5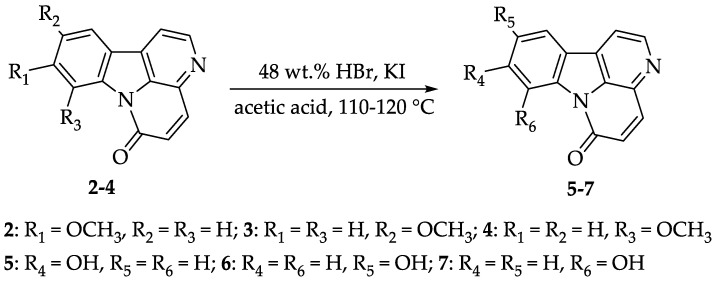 Figure 6
Figure 6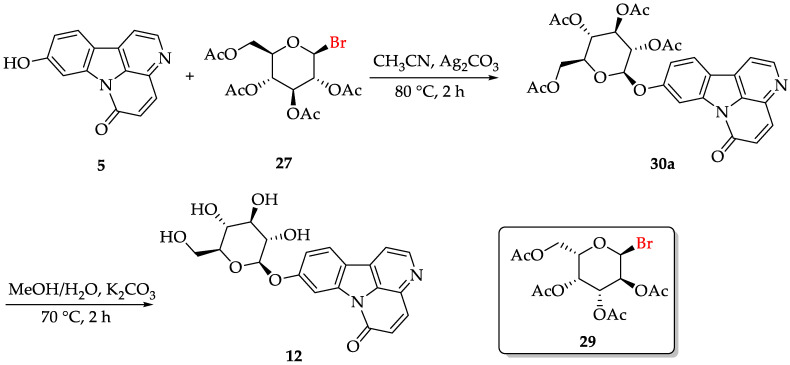 Figure 7
Figure 7- —National Natural Science Foundation of China
- —Natural Science Foundation of Guangxi
- —Research Start-up Foundation of Guangxi Minzu University
- —Natural Science Foundation of the Educational Department of Guangdong Province
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPhytochemical compounds biological activities · Traditional and Medicinal Uses of Annonaceae · Biochemical and Structural Characterization
1. Introduction
In recent decades, there has been a renewed focus on natural products for innovative drug discovery [1]. Alkaloids, a class of nitrogen-containing organic compounds found in plants, fungi, bacteria and other organisms, have been used therapeutically for over 3000 years [2,3]. Many alkaloids, including paclitaxel, vincristine, atropine, pilocarpine, quinine and vincamine, are used clinically [4]. The canthin-6-one (or canthinone) alkaloids are a subclass of β-carboline alkaloids with an additional D-ring (Figure 1). Canthin-6-one 1 was first isolated in 1952 by Haynes from the Australian plant species Pentaceras australis (F.Muell.) Benth. (Rutaceae) [5]. Since then, nearly 90 canthin-6-one alkaloids have been isolated from over fifty species (the structure of some canthin-6-one alkaloids is shown in Figure 1). These alkaloids are predominantly found in the root, bark and leaves of Rutaceae and Simaroubaceae, with smaller quantities present in their heartwood. Additionally, genera from Malvaceae, Amaranthaceae, Zygophyllaceae, Caryophylaceae and Leitneriaceae also contain canthin-6-one alkaloids [6].
Canthin-6-one alkaloids and their derivatives demonstrate a wide range of biological activities, including antiviral [7,8,9], antitumor [10,11,12], antiparasitic [13,14,15], anti-inflammatory [16,17], antibacterial [18,19,20] and antifungal [21,22]. Notably, canthine-6-one alkaloids and their derivatives have gained significant attention due to their antimicrobial potencies. For example, O’Donnell et al. isolated two canthin-6-ones, with minimum inhibitory concentrations (MIC) ranging from 8 to 32 µg/mL against a panel of fast-growing Mycobacterium species and 8 to 64 µg/mL against multidrug-resistant and methicillin-resistant strains of Staphylococcus aureus [20]. Wang et al. synthesized a novel series of hydrophilic derivatives of canthin-6-one with a peak MIC of 0.98 µg/mL against S. aureus [18,23,24]. Li et al. synthesized twenty-two new 3-N-benzylated 10-methoxy canthin-6-ones through a quaternization reaction. The in vitro antibacterial activity against R. solanacearum, P. syringae and B. cereus revealed that two compounds demonstrated 8-fold superior activities (MIC = 3.91 μg/mL) against the agricultural pathogenic bacteria R. solanacearum and P. syringae, compared to agrochemical streptomycin sulfate. They showed good antibacterial activity against B. cereus, along with favorable “drug-like” properties, low cytotoxicity and no adverse effects on seed germination [25]. Dai et al. synthesized C2- and C5-modified, D-ring-rearranged canthin-6-ones, along with C10 and truncated canthin-6-ones in two separate studies, to evaluate their antibacterial activity and SARs [26,27]. The C_2_-substituted and D-ring-rearranged compound exhibited broad-spectrum antibacterial activity, effective against all the tested bacteria, including S. aureus, B. subtilis (MIC = 12.5 nmol/mL), MRSA, B. cereus and R. solanacearum (MIC = 25 nmol/mL), but not E. coli (MIC > 200 nmol/mL) [26]. Additionally, the C-ring-truncated canthin-6-one showed a broad-spectrum antibacterial effect, with an eight-fold superior activity against S. aureus. It showed a two-fold superiority against Ralstonia solanacearum compared to fosfomycin sodium and propineb, with an MIC of 2 μg/mL.
Due to the promising applications of canthin-6-one alkaloids, numerous reports have been published on their synthesis. In 2022, Cebrián-Torrejón’s group evaluated a series of synthesis strategies for canthin-6-one scaffolds (Figure 2) [6]. Most of these strategies employed tryptamine or indole-based precursors, primarily achieved through reductive amination, Pictet–Spengler reactions, amidation reactions, catalytic hydrogen transfer and catalytic oxidation reactions. However, it is found that some of the original synthetic approaches do not rely on tryptamine or its derivatives. These methods include the following: the C_10_N_2_ + C_2_ + C_2_ strategy, which was used to synthesize compounds 8 and 9; the C_10_N_2_ + C_4_ strategy, which was used to synthesize compounds 1, 8 and 10; the C_8_N_2_ + C_6_ strategy, which was used to synthesize compound 1; and the C_12_N_2_ + C_2_ strategy, which led to the synthesis of canthine alkaloid 11. Among these strategies, the C_8_N_2_ + C_6_ strategy is the most prominent, with canthin-6-one 1 synthesized in three steps, achieving a total yield of 86.4% [6]. The strategies mentioned above were used to structure and guide our synthesis approach in the present study.
So far, there are no reports on the synthesis of some canthin-6-one alkaloids, including canthin-6-ones 4, 5, 7, 12 and 13. There is limited literature available on the bioactivities of canthin-6-one alkaloids and their potential antimicrobial activities. Herein, we report the synthesis and evaluation of the antimicrobial activity of canthin-6-one alkaloids, along with a novel series of canthin-6-one alkaloids’ derivatives for the first time.
2. Results and Discussion
2.1. Synthesis of Canthin-6-Ones 1-4
A variety of synthetic strategies for constructing the canthin-6-one skeleton have been reported [6]. For instance, the canthin-6-one skeleton can be constructed through the cleavage of either the C1-C14 and C15-C16 bonds or the C6-N and C15-C16 bonds [21,28]. By comparing different synthetic stragegies, the retrosynthetic analysis of canthin-6-one skeleton in this study was based on reported synthetic routes for canthin-6-one alkaloids [29], and is illustrated in Figure 3. This approach primarily focused on constructing ring B. Intermediate 14 was generated by cleaving the amide bond at C13, while building blocks 16 and 17 were derived from intermediate 15 through the cleavage of the C12-C14 bond. These two bond formations were achieved via an intermolecular C-C coupling, followed by an intramolecular C-N coupling. Notably, incorporating ring A in the final stage of synthesis provided structural flexibility, allowing for modification at the C8-C11 positions.
To synthesize canthin-6-ones 1-4, the key intermediate 16 was initially prepared via a three-step reaction sequence, starting with commercially available 6-methoxypyridin-3-amine 18 as the precursor (Scheme 1), as described in the reference [30]. Briefly, the first step involved the condensation of 18 with Meldrum’s acid and trimethoxymethane. This domino sequence included an addition-elimination reaction, resulting in the enamine intermediate 19 with a yield of 74.1%. In the next step, thermal Friedel–Crafts cyclization with decarboxylation was used to synthesize 20, and it had a good yield of 73.3%. Finally, a straightforward bromination of 20 with PBr_3_ at 0 °C yielded the key intermediate 16, which had a yield of 73.3% after purification by silica gel column chromatography. Compound 16 was obtained in three steps with an overall yield of 44.5%, as reported in the reference [30]. Using the same method, we achieved an overall yield of 39.8%. The reduction in overall yield is attributed to the purification of our final product by column chromatography.
Subsequently, compound 21 was prepared from intermediate 16 and 2-chlorophenylboronic acid vis Suzuki–Miyaura coupling, achieving an excellent yield (89.9–99.5%) when Pd(dppf)Cl_2_ was used together with K_2_CO_3_ in a dioxane/H_2_O solvent system. Refluxing 21 with aqueous HCl in dioxane resulted in the formation of naphthyridone 22. During this step, some of product 22 may have formed a salt under acidic conditions, which led to a significant product loss when a direct extraction of the aqueous phase was carried out with CH_2_Cl_2_. To address this, we adjusted the pH to 7-8 using solid Na_2_CO_3_ before extraction, which resulted in a high-yield recovery of product 22 (82.2-97.7%). The NMR data for intermediates 21a, 21b, 22a and 22b were consistent with those of the corresponding intermediates reported in reference [29]. Intermediates 21c and 21d were obtained for the first time, along with 22c and 22d, and their structures were confirmed by NMR spectroscopy. The final C-N coupling was carried out under Buchwald’s conditions (CuI, DMEDA as the ligand and H_2_O in dioxane at reflux), finishing in 1.5-2.0 h with a yield of 90.9-98.9%. Therefore, it is evident that the approach successfully synthesized canthin-6-ones 1-4 in three steps from commercially available starting material 16 with high yields (Scheme 1) [29].
2.2. Synthesis of Compounds 27-28
The synthesis of glycoside alkaloids 12 and 13 was planned using Koenig–Knorr glycosylation. Therefore, 2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl bromide 27 needs to be synthesized first. β-D-glucose 23 was esterified followed by bromination to yield 27 [31], with a total yield of 49.0%, as described in Scheme 2. For the synthesis of the analogs of glycoside alkaloids 12, acetobromo-β-D-maltose 28 was obtained using the same synthetic method [32], with commercially available aceto-β-D-maltose 26 as raw material (yield: 76.2%).
2.3. Synthesis of Compounds 5-7, 12-13 and 30-31
Zhao et al. reported that deprotecting the methoxy group of 10-methoxycanthin-6-one 3 using boron tribromide can yield 10-hydroxycanthin-6-one 6 [24]. However, this reaction requires a temperature of −78 °C, and the yield is moderate. To make the operation more convenient and achieve a higher yield, we explored a strategy for the methoxy-to-hydroxyl transformation. A widely used method for deprotecting the methoxy group involves 48 wt.% HBr. We successfully applied this method to synthesize 5-7, and compounds 5 and 7 were obtained for the first time (Scheme 3). Using potassium iodide as a catalyst and adding a controlled amount of acetic acid under heating, the yields of alkaloids 5-7 ranged from 82.9 to 99.1%. An HPLC purity test was conducted for natural product 5 (Figure S1), confirming a purity of over 98%.
With 5-7 in hand, glucopyranoside alkaloids and their derivatives were possible to synthesize (the names of derivatives are shown in Table 1). When canthin-6-one 5 reacted with bromide 27 in the presence of silver carbonate, Koenig–Knorr glycosylation occurred, yielding 30a with an 83.5% yield. Subsequent hydrolysis with potassium carbonate led to the synthesis of glucopyranoside alkaloid 12 (with a yield of 86.6%) (Scheme 4). When the starting material is canthin-6-one 6, the same method yields glucopyranoside alkaloid 13. Glucopyranoside alkaloids 12 and 13 were synthesized in six steps, with overall yields of 61.5% and 41.2%, respectively, starting from commercially available starting material 16. Similarly, when canthin-6-one 5 and 6 and commercially available available 2,3,4,6-Tetra-O-acetyl-α-D-galactopyranosyl bromide 29 or canthin-6-one 6 and bromide 28 are used as the starting material, analogs 31a-31c can be obtained with good yields (yield range: 75.6–94.1%). Unfortunately, compound 7 did not react with compound 29, likely due to the steric hindrance of the carbonyl group, which was relatively large and positioned close to the C8 site, preventing the glycosylation reaction from occurring.
2.4. Comparison of NMR Data
By comparing the NMR spectroscopic data, it was confirmed that the synthesized compounds 1-7 and 12-13 are natural canthin-6-ones 1-7 and 12-13. For example, the ^13^C NMR chemical shifts of natural and synthetic compound 12 are presented in Table 2. The ^13^C NMR of canthin-6-one 12 closely matched that of the synthetic compound 12, with the largest significant discrepancies of 0.2 ppm observed for some carbon atoms. This strongly supports the identification of synthetic compound 12 as the natural product of canthin-6-one 12, as reported by Yamasaki et al. [33].
2.5. Initial Screening for Antimicrobial Activity
The synthesized compounds were initially evaluated against four microbial human pathogen strains (C. albicans, C. neoformans, S. aureus and E. coli) using the micro-dilution method. The results are summarized in Table 3.
As shown in Table 3, canthin-6-ones 1 and 7 demonstrated inhibition rates exceeding 80% against C. albicans, while canthin-6-ones 1, 2, 4 and 7 showed more than 70% inhibition against C. neoformans at 50 µg/mL, comparable to the positive control drug Ketoconazole. Among them, canthin-6-one 1 displayed the highest activity, with inhibition rates of 95.7% against C. albicans and 96.9% against C. neoformans. Canthin-6-ones 1-3 and 7 showed activity against S. aureus, with inhibition rates ranging from 88.1% to 99.8%. However, all tested compounds exhibited inhibition rates below 50% against E. coli at the same concentration, indicating a lack of antibacterial activity against this strain. Additionally, all derivatives showed no inhibitory effect against the four microbial phytopathogens.
The antimicrobial activity of canthin-6-ones 2, 4-5 and 12-13 against these four microbial human pathogens was reported here for the first time, along with the antimicrobial activity of canthin-6-ones 2 and 4-5 against C. albicans, C. neoformans and E. coli.
3. Materials and Methods
3.1. General Chemical Procedures
All chemicals were purchased from commercial suppliers and used without further purification. All reagents and solvents were reagent or HPLC grade. ^1^H and ^13^C NMR spectra were recorded on a Bruker spectrometer at 400 MHz (Billerica, MA, USA) in the indicated solvents. Chemical shifts (δ) were expressed in ppm with reference to the solvent signals (CDCl_3_, ^1^H: 7.26 ppm; ^13^C: 77.00 ppm), and coupling constants (J) are reported in Hz. Standard acronyms representing multiplicity are used as follows: brs = broad singlet, s = singlet, d = doublet, t = triplet and m = multiplet. All NMR experiments were obtained using standard pulse sequences supplied by the vendor. Thin-layer chromatography (TLC) was performed on thin-layer chromatography GF 254 (10–40 μm) silica gel plates. Column chromatography was carried out on silica gel (200–300 mesh, Qingdao Marine Chemical Factory, Qingdao, China). LC-MS instrument was purchased from Agilent Technology (Hong Kong, China) Co., Ltd. (Agilent 1260 + 6125).
3.2. Synthesis of Target Compounds
3.2.1. Synthesis of Compound 19 [30]
A mixture of 2,2-dimethyl-1,3-dioxane-4,6-dione (2.9 g, 20.0 mmol) and trimethoxymethane (19.1 g, 180.0 mmol) in ethanol (6 mL) was heated under reflux for 2 h. Afterwards, 6-methoxypyridin-3-amine (2.5 g, 20.0 mmol) was added, and the resulting solution was stirred under reflux for 12 additional hours. Upon cooling to room temperature, the mixture was diluted with hexane (30 mL) and stirred in an ice bath for 5–10 min, resulting in the formation of a significant amount of precipitate. The precipitate was filtered and washed with hexane, yielding the product (74.1% yield) as a purple solid. ^1^H NMR (400 MHz, CDCl_3_) δ (ppm): 11.17 (d, J = 14.20 Hz, 1H), 8.48 (d, J = 14.24 Hz, 1H), 8.11 (d, J = 2.88 Hz, 1H), 7.52 (dd, J = 8.92, 2.92 Hz, 1H), 6.82 (d, J = 8.92 Hz, 1H), 3.94 (s, 3H), 1.75 (s, 6H).
3.2.2. Synthesis of Compound 20 [30]
Dowtherm A (approximately 3:7 diphenyl/diphenyl oxide, 40 mL) was added to a single-neck round-bottom flask equipped with a thermocouple and a funnel, and the solvent was heated to 220 °C. To the heated Dowtherm A, compound 19 (4.1 g, 14.7 mmol) was added over a 30 min period, maintaining the temperature at approximately 220 °C. The funnel was then rinsed with 10 mL of Dowtherm A. The reaction mixture was heated for an additional 10 min, after which the heat source was removed. The resulting suspension was allowed to cool to room temperature and treated with n-hexane (50 mL). The mixture was stirred in an ice bath for 5–10 min, and the solid product was filtered, washed with hexane (50 mL) and dried in vacuo to yield 1.9 g of compound 20 as a tan solid (73.3%). The crude product was used directly for the next step of the reaction. ^1^H NMR (400 MHz, DMSO-d_6_) δ (ppm): 11.87 (br s, 1H), 7.95 (br s, 2H), 7.16 (d, J = 8.96 Hz, 1H), 6.22 (br s, 1H), 3.93 (s, 3H).
3.2.3. Synthesis of Compound 16 [30]
PBr_3_ (1.1 mL) was added to a solution of compound 20 (1.8 g, 10.2 mmol) in DMF (2.1 mL) under a nitrogen atmosphere at 0 °C. The mixture was stirred at 0 °C for 1 h, then quenched with H_2_O (107 mL) and 6 N NaOH (0.28 mL), followed by extraction with ethyl acetate (3 × 100 mL). The combined organic layers were washed with brine (50 mL) and dried over anhydrous Na_2_SO_4_, and the solvent was removed under vacuum. The residue was purified by flash column chromatography to obtain a white solid product with 73.3% yield. ^1^H NMR (400 MHz, DMSO-d_6_) δ (ppm): 8.59 (d, J = 4.76 Hz, 1H), 8.29 (d, J = 8.96 Hz, 1H), 8.07 (d, J = 4.68 Hz, 1H), 7.33 (d, J = 9.00 Hz, 1H), 4.06 (s, 3H).
3.2.4. Synthesis of Compounds 21
To a stirred solution of compound 16 (0.3 g, 1.3 mmol), a 3:1 mixture of 1,4-dioxane/H_2_O (2.5 mL), K_2_CO_3_ (0.3 g, 2.5 mmol), Pd(dppf)Cl_2_ (36.9 mg, 0.05 mmol) and benzene boric acid (1.9 mmol) was added. The reaction mixture was warmed to 100–105 °C and stirred for 2–3 h. Upon completion, the mixture was diluted with H_2_O (20 mL) and extracted with CH_2_Cl_2_ (3 × 20 mL). The combined organic layers were washed with brine (40 mL) and dried over anhydrous Na_2_SO_4_, and the solvent was removed under vacuum. The residue was purified by flash column chromatography to obtain the final product.
Compound 21a: colorless transparent oil, yield: 89.9%. ^1^H NMR (400 MHz, CDCl_3_) δ (ppm): 8.83 (d, J = 4.44 Hz, 1H), 8.25 (d, J = 9.04 Hz, 1H), 7.55–7.50 (m, 2H), 7.44–7.40 (m, 1H), 7.39–7.33 (m, 2H), 7.12 (d, J = 9.04 Hz, 1H), 3.81 (s, 3H). ^13^C NMR (100 MHz, CDCl_3_) δ (ppm): 161.9, 147.5, 144.7, 142.3, 140.2, 140.0, 136.6, 133.7, 131.9, 129.5, 129.4, 126.3, 125.0, 116.7, 53.7.
Compound 21b: white solid, yield: 90.1%. ^1^H NMR (400 MHz, DMSO-d_6_) δ (ppm): 8.84 (d, J = 4.52 Hz, 1H), 8.32 (d, J = 9.00 Hz, 1H), 7.62 (d, J = 4.44 Hz, 1H), 7.45 (d, J = 8.48 Hz, 1H), 7.26 (d, J = 9.00 Hz, 1H), 7.20 (d, J = 2.52 Hz, 1H), 7.05 (dd, J = 8.60, 2.56 Hz, 1H), 3.85 (s, 3H), 3.77 (s, 3H). ^13^C NMR (100 MHz, DMSO-d_6_) δ (ppm): 161.2, 159.8, 147.7, 143.4, 141.7, 140.4, 139.3, 133.2, 132.8, 127.9, 125.4, 116.4, 114.5, 112.8, 55.7, 53.2.
Compound 21c: yellow transparent oil, yield: 98. 9%. ^1^H NMR (400 MHz, DMSO-d_6_) δ (ppm): 8.86 (d, J = 4.44 Hz, 1H), 8.33 (d, J = 9.00 Hz, 1H), 7.65 (d, J = 4.44 Hz, 1H), 7.50 (d, J = 8.28 Hz, 1H), 7.27 (d, J = 9.04 Hz, 1H), 7.11–7.03 (m, 2H), 3.79 (s, 3H), 3.76 (s, 3H). ^13^C NMR (100 MHz, DMSO-d_6_) δ (ppm): 161.3, 157.6, 147.7, 143.6, 141.6, 140.4, 139.0, 136.8, 129.9, 125.1, 123.8, 117.0, 116.5, 115.6, 55.6, 53.2.
Compound 21d: yellow transparent oil, yield: 99.0%. ^1^H NMR (400 MHz, DMSO-d_6_) δ (ppm): 8.85 (d, J = 4.40 Hz, 1H), 8.33 (d, J = 9.04 Hz, 1H), 7.60 (d, J = 4.40 Hz, 1H), 7.42 (t, J = 8.04 Hz, 1H), 7.29–7.22 (m, 2H), 7.05 (dd, J = 7.64, 1.36 Hz, 1H), 3.92 (s, 3H), 3.72 (s, 3H). ^13^C NMR (100 MHz, DMSO-d_6_) δ (ppm): 161.3, 154.7, 147.7, 143.8, 141.6, 140.4, 139.1, 137.4, 127.2, 125.0, 123.3, 120.8, 116.5, 112.2, 56.3, 53.1.
3.2.5. Synthesis of Compounds 22
To a stirred solution of compound 21 (1.5 mmol) in 1,4-dioxane (7.5 mL), a 1:1 mixture of HCl and H_2_O (10.5 mL) was added. The reaction mixture was heated to 100–105 °C and stirred for 2–3 h. Upon completion, H_2_O (30 mL) was added, and the pH was adjusted to 7 by the addition of solid Na_2_CO_3_. The mixture was then extracted with CH_2_Cl_2_ (3 × 30 mL). The combined organic layers were washed with brine (30 mL) and dried over anhydrous Na_2_SO_4_, and the solvent was removed under vacuum. The crude product was obtained directly for the next step of the reaction.
Compound 22a: white solid, yield: 97.7%. ^1^H NMR (400 MHz, CDCl_3_) δ (ppm): 8.61 (d, J = 4.68 Hz, 1H), 8.45 (br s, 1H), 8.04 (d, J = 9.80 Hz, 1H), 7.59 (dd, J = 7.84, 1.40 Hz, 1H), 7.53–7.42 (m, 2H), 7.34–7.28 (m, 2H), 6.85 (d, J = 9.80 Hz, 1H). ^13^C NMR (100 MHz, CDCl_3_) δ (ppm): 161.7, 145.1, 142.5, 137.9, 133.5, 132.3, 132.1, 131.4, 131.2, 130.8, 128.0, 126.0, 125.5.
Compound 22b: white solid, yield: 96.3%. ^1^H NMR (400 MHz, DMSO-d_6_) δ (ppm): 10.97 (br s, 1H), 8.51 (d, J = 4.64 Hz, 1H), 7.99 (d, J = 9.72 Hz, 1H), 7.33 (d, J = 1.36 Hz, 1H), 7.32 (d, J = 5.28 Hz, 1H), 7.20 (d, J = 2.48 Hz, 1H), 7.04 (dd, J = 8.56, 2.52 Hz, 1H), 6.76 (d, J = 9.76 Hz, 1H), 3.85 (s, 3H). ^13^C NMR (100 MHz, DMSO-d_6_) δ (ppm): 161.6, 160.6, 144,1, 141.5, 137.2, 133.7, 133.5, 133.3, 132.2, 126.2, 126.0, 125.5, 115.1, 113.9, 55.8.
Compound 22c: yellow solid, yield: 82.2%. ^1^H NMR (400 MHz, DMSO-d_6_) δ (ppm): 10.98 (br s, 1H), 8.53 (d, J = 4.64 Hz, 1H), 8.00 (d, J = 9.72 Hz, 1H), 7.49 (d, J = 8.88 Hz, 1H), 7.37 (d, J = 4.60 Hz, 1H), 7.08 (dd, J = 8.88, 3.08 Hz, 1H), 7.00 (d, J = 3.08 Hz, 1H), 6.78 (d, J = 9.76 Hz, 1H), 3.78 (s, 3H). ^13^C NMR (100 MHz, DMSO-d_6_) δ (ppm): 161.6, 158.3, 144.1, 141.4, 137.3, 134.1, 133.0, 130.6, 126.1, 125.7, 123.6, 116.8, 116.6, 55.6.
Compound 22d: pink solid, yield: 96.1%. ^1^H NMR (400 MHz, DMSO-d_6_) δ (ppm): 10.97 (br s, 1H), 8.52 (d, J = 4.64 Hz, 1H), 8.00 (d, J = 9.80 Hz, 1H), 7.41 (t, J = 7.96 Hz, 1H), 7.33 (d, J = 4.64 Hz, 1H), 7.25 (dd, J = 8.40, 1.40 Hz, 1H), 6.95 (dd, J = 7.60, 1.40 Hz, 1H), 6.77 (d, J = 9.76 Hz, 1H), 3.92 (s, 3H). ^13^C NMR (100 MHz, DMSO-d_6_) δ (ppm): 161.6, 155.2, 144.2, 141.5, 137.2, 134.7, 133.9, 133.0, 128.2, 126.1, 125.6, 122.7, 121.0, 113.1, 56.3.
3.2.6. Synthesis of Compounds 1-4
To a stirred solution of compound 22 (1.0 mmol) in 1,4-dioxane (4.0 mL), Cs_2_CO_3_ (651.6 mg, 2.0 mmol), CuI (19.1 mg, 0.1 mmol), DMEDA (17.6 mg) and H_2_O (36.0 mg) were sequentially added. The reaction mixture was heated to 100–105 °C and stirred for 1.5–2 h. Upon completion, the mixture was diluted with H_2_O (20 mL) and extracted with CH_2_Cl_2_ (3 × 20 mL). The combined organic layers were washed with brine (30 mL) and dried over anhydrous Na_2_SO_4_, and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography to afford the title products.
Compound 1: pale yellow solid, yield: 90.9%. ^1^H NMR (400 MHz, CDCl_3_) δ (ppm): 8.80 (d, J = 5.08 Hz, 1H), 8.64 (d, J = 8.20 Hz, 1H), 8.08 (d, J = 7.72 Hz, 1H), 8.00 (d, J = 9.80 Hz, 1H), 7.93 (d, J = 4.96 Hz, 1H), 7.68 (ddd, J =8.20, 7.24, 1.20 Hz, 1H), 7.50 (td, J = 7.84, 1.08 Hz, 1H), 6.96 (d, J = 9.76 Hz, 1H). ^13^C NMR (100 MHz, CDCl_3_) δ (ppm): 159.7, 146.0, 139.8, 139.6, 136.4, 132.2, 131.0, 130.4, 129.1, 125.8, 124.5, 122.8, 117.4, 116.5. MS m/z 243.0 [M + Na]^+^ (calcd for C_14_H_8_N_2_O, 220.06).
Compound 2: pale yellow solid, yield: 98.9%. ^1^H NMR (400 MHz, CDCl_3_) δ (ppm): 8.74 (d, J = 5.00 Hz, 1H), 8.15 (d, J = 2.40 Hz, 1H), 7.98 (d, J = 9.76 Hz, 1H), 7.90 (d, J = 8.64 Hz, 1H), 7.80 (d, J = 5.00 Hz, 1H), 7.04 (dd, J = 8.64, 2.40 Hz, 1H), 6.93 (d, J = 9.80 Hz, 1H), 3.97 (s, 3H). ^13^C NMR (100 MHz, CDCl_3_) δ (ppm): 162.6, 159.8, 146.1, 141.3, 140.0, 135.8, 132.4, 130.5, 128.6, 123.5, 117.3, 115.7, 114.3, 101.4, 56.1. MS m/z 273.1 [M + Na]^+^ (calcd for C_15_H_10_N_2_O_2_, 250.07).
Compound 3: bright yellow solid, yield: 92.0%. ^1^H NMR (400 MHz, CDCl_3_) δ (ppm): 8.78 (d, J = 5.00 Hz, 1H), 8.50 (d, J = 8.92 Hz, 1H), 7.98 (d, J = 9.80 Hz, 1H), 7.89 (d, J = 5.00 Hz, 1H), 7.51 (d, J = 2.52 Hz, 1H), 7.22 (dd, J = 8.96, 2.52 Hz, 1H), 6.95 (d, J = 9.76 Hz, 1H), 3.94 (s, 3H). ^13^C NMR (100 MHz, CDCl_3_) δ (ppm): 159.3, 158.0, 145.6, 139.2, 136.3, 133.9, 132.5, 130.5, 129.2, 125.7, 118.1, 118.1, 116.5, 106.6, 56.1. MS m/z 251.1 [M + H]^+^ (calcd for C_15_H_10_N_2_O_2_, 250.07).
Compound 4: bright yellow solid, yield: 95.0%. ^1^H NMR (400 MHz, CDCl_3_) δ (ppm): 8.74 (d, J = 5.28 Hz, 1H), 7.92 (d, J = 9.72 Hz, 1H), 7.89 (d, J = 4.96 Hz, 1H), 7.67 (dd, J = 7.72, 1.04 Hz, 1H), 7.45 (t, J = 7.92 Hz, 1H), 7.23 (d, J = 8.08 Hz, 1H), 6.96 (d, J = 9.64 Hz, 1H), 4.07 (s, 3H). ^13^C NMR (100 MHz, CDCl_3_) δ (ppm): 158.0, 149.8, 145.2, 138.8, 137.1, 130.7, 130.3, 129.3, 127.5, 127.1, 115.8, 115.2, 114.8, 57.5. MS m/z 273.0 [M + Na]^+^ (calcd for C_15_H_10_N_2_O_2_, 250.07).
3.2.7. Synthesis of Compound 25 [31]
To a gently refluxing solution, Ac_2_O (14.2 g, 139.0 mmol) containing NaOAc (0.6 g, 7.0 mmol) and powdered D-glucose 23 (2.5 g, 13.9 mmol) were slowly added over a period of 15 min. The mixture was then heated in reflux for additional 5 min before being cooled to room temperature. The reaction was quenched by addition of ice under sonication, during which compound 25 was precipitated. The resulting white powder was filtered and washed with H_2_O until the acetic acid odor was removed, then dried under vacuum to yield the desired product as a white powder (4.2 g, 77.5% yield). ^1^H NMR (400 MHz, MeOD) δ (ppm): 5.82 (d, J = 8.32 Hz, 1H), 5.35 (t, J = 9.48 Hz, 1H), 5.11–5.01 (m, 2H), 4.28 (dd, J = 12.44, 4.52 Hz, 1H), 4.11 (dd, J = 12.48, 2.28 Hz, 1H), 4.05–3.99 (m, 1H), 2.08 (s, 3H), 2.05 (s, 3H), 2.02 (s, 6H), 1.98 (s, 3H).
3.2.8. Synthesis of Compounds 27 and 28 [32]
To a solution of 25 or 26 (5.0 mmol) in CH_2_Cl_2_ (7.0 mL) cooled to 0 °C in an ice-water bath, 33% HBr in acetic acid solution (1.24 mL) was added dropwise. The mixture was gradually warmed to 25 °C and stirred for 1.5 h. The reaction was quenched by adding saturated Na_2_CO_3_ solution, and the mixture was extracted with CH_2_Cl_2_. The solvent was removed under reduced pressure, and the residue was purified by flash column chromatography to afford the desired products.
Compound 27: white solid, yield: 63.2%. ^1^H NMR (400 MHz, DMSO-d_6_) δ (ppm): 5.40–5.05 (m, 2H), 4.96–4.76 (m, 2H), 4.72–4.64 (m, 1H), 4.13–4.08 (m, 1H), 4.02–3.97 (m, 1H), 2.00 (t, J = 1.12 Hz, 3H), 1.98–1.95 (m, 6H), 1.93 (d, J = 12.80 Hz, 3H).
Compound 28: white solid, yield: 76.2%. ^1^H NMR (400 MHz, DMSO-d_6_) δ (ppm): 6.82 (d, J = 3.76 Hz, 1H), 5.42 (dd, J = 9.84, 8.56 Hz, 1H), 5.28 (d, J = 3.96 Hz, 1H), 5.26–5.19 (m, 1H), 5.04–4.96 (m, 1H), 4.91 (d, J = 3.84 Hz, 1H), 4.88 (dd, J = 3.80, 1.16 Hz, 1H), 4.42 (dd, J = 12.96, 2.36 Hz, 1H), 4.26–4.15 (m, 4H), 4.05–4.00 (m, 2H), 2.07 (s, 3H), 2.02 (s, 3H), 2.01 (s, 3H), 2.00 (s, 3H), 1.99 (s, 3H), 1.98 (s, 3H), 1.96 (s, 3H).
3.2.9. Synthesis of Compounds 5-7
To a stirred solution of compound 2 (62.6 mg, 0.25 mmol) and KI (4.2 mg, 0.025 mmol) in 48 wt.% HBr (0.85 mL, 7.5 mmol) under a nitrogen atmosphere, acetic acid (0.12 mL) was slowly added. The mixture was warmed to 110–120 °C and stirred overnight. At the end of the reaction, H_2_O (10.0 mL) was added, and the pH was adjusted to 7–8 by addition of solid Na_2_CO_3_. The solid was isolated by suction filtration, and the filter cake was washed sequentially with cold water and CH_2_Cl_2_. The solid was then dried under vacuum to obtain the desired products: 5-7.
Compound 5: dark yellow solid, yield: 99.1%. ^1^H NMR (400 MHz, DMSO-d_6_) δ (ppm): 10.71 (br s, 1H), 8.82 (d, J = 5.48 Hz, 1H), 8.28 (d, J = 5.44 Hz, 1H), 8.18 (d, J = 8.56 Hz, 1H), 8.11 (d, J = 9.84 Hz, 1H), 7.88 (d, J = 2.20 Hz, 1H), 7.03 (d, J = 9.84 Hz, 1H), 6.99 (dd, J = 8.60, 2.24 Hz, 1H). ^13^C NMR (100 MHz, DMSO-d_6_) δ (ppm): 161.8, 158.4, 142.5, 141.7, 136.4, 132.4, 131.9, 131.8, 130.1, 125.6, 116.2, 114.9, 114.6, 102.9. MS m/z 237.0 [M + H]^+^ (calcd for C_14_H_8_N_2_O_2_, 236.06).
Compound 6: orange yellow solid, yield: 82.9%. ^1^H NMR (400 MHz, DMSO-d_6_) δ (ppm): 9.94 (br s, 1H), 8.78 (d, J = 5.04 Hz, 1H), 8.27 (d, J = 8.80 Hz, 1H), 8.23 (d, J = 5.04 Hz, 1H), 8.06 (d, J = 9.76 Hz, 1H), 7.65 (d, J = 2.48 Hz, 1H), 7.16 (dd, J = 8.72, 2.44 Hz, 1H), 6.94 (d, J = 9.72 Hz, 1H). ^13^C NMR (100 MHz, DMSO-d_6_) δ (ppm): 158.4, 155.7, 145.4, 139.0, 135.5, 132.0, 131.8, 129.7, 128.7, 125.5, 118.6, 117.3, 117.0, 109.1. MS m/z 235.1 [M − H]^−^ (calcd for C_14_H_8_N_2_O_2_, 236.06).
Compound 7: yellow solid, yield: 85.5%. ^1^H NMR (400 MHz, DMSO-d_6_) δ (ppm): 11.91 (s, 1H), 8.86 (d, J = 4.88 Hz, 1H), 8.26 (d, J = 4.36 Hz, 1H), 8.24 (s, 1H), 7.72 (d, J = 7.52 Hz, 1H), 7.42 (t, J = 7.88 Hz, 1H), 7.13–7.07 (m, 2H). ^13^C NMR (100 MHz, DMSO-d_6_) δ (ppm): 160.4, 153.8, 147.2, 145.9, 141.5, 136.0, 131.0, 128.6, 127.0, 126.8, 126.8, 118.5, 117.8, 114.0. MS m/z 259.1 [M + Na]^+^ (calcd for C_14_H_8_N_2_O_2_, 236.06).
3.2.10. Synthesis of Compounds 30
Compound 7 (0.10 mmol) was dissolved in anhydrous CH_3_CN (2.5 mL), and Ag_2_CO_3_ (68.9 mg, 0.25 mmol) was added. The mixture was heated until it boiled, followed by the addition of a solution of 27, 28 or 29 (0.30 mmol). The reaction was then stirred and heated under azeotropic conditions for an additional 2–3 h. Afterwards, the inorganic salts were removed by filtration, and the solvent was evaporated from the filtrate, leaving a dry residue. The product was purified by silica gel column chromatography using a stepwise gradient of PE/EtOAc (2:1 to 1:2), yielding a pure product: 30.
Compound 30a: brown solid, yield: 83.5%. ^1^H NMR (400 MHz, DMSO-d_6_) δ (ppm): 8.81 (d, J = 5.00 Hz, 1H), 8.35 (d, J = 8.64 Hz, 1H), 8.24 (d, J = 5.00 Hz, 1H), 8.16–8.14 (m, 1H), 8.12 (d, J = 4.16 Hz, 1H), 7.25 (dd, J = 8.64, 2.36 Hz, 1H), 6.99 (d, J = 9.88 Hz, 1H), 5.80 (d, J = 7.88 Hz, 1H), 5.51 (t, J = 9.56 Hz, 1H), 5.16 (dd, J = 9.76, 7.88 Hz, 1H), 5.05 (t, J = 9.72 Hz, 1H), 4.39–4.32 (m, 1H), 4.23 (dd, J = 12.36, 5.72 Hz, 1H), 4.11 (dd, J = 12.32, 2.40 Hz, 1H), 2.06 (s, 3H), 2.03 (s, 6H), 1.99 (s, 3H). ^13^C NMR (100 MHz, DMSO-d_6_) δ (ppm): 170.1, 169.6, 169.4, 169.2, 159.0, 158.4, 146.1, 140.1, 140.1, 135.5, 132.1, 130.0, 128.4, 124.8, 119.3, 116.9, 114.6, 104.6, 97.5, 71.9, 71.1, 70.8, 68.1, 61.7, 20.5, 20.4, 20.4, 20.3. MS m/z 589.4 [M + Na]^+^ (calcd for C_28_H_26_N_2_O_11_, 566.15).
Compound 30b: brown solid, yield: 87.7%. ^1^H NMR (400 MHz, DMSO-d_6_) δ (ppm): 8.78 (d, J = 5.00 Hz, 1H), 8.23 (d, J = 4.96 Hz, 1H), 8.09 (d, J = 4.48 Hz, 1H), 7.66 (d, J = 1.96 Hz, 1H), 7.37–7.34 (m, 1H), 6.96 (t, J = 10.04 Hz, 2H), 5.70 (d, J = 8.00 Hz, 1H), 5.45 (t, J = 9.56 Hz, 1H), 5.16 (dd, J = 9.76, 7.96 Hz, 1H), 5.09–5.04 (m, 1H), 4.25 (dd, J = 12.28, 5.20 Hz, 1H), 4.18 (d, J = 2.44, 1H), 4.15 (d, J = 2.40, 1H), 2.09 (s, 3H), 2.03 (s, 3H), 2.01 (s, 3H), 1.99 (s, 3H). ^13^C NMR (100 MHz, DMSO-d_6_) δ (ppm): 170.0, 169.6, 169.3, 169.2, 158.4, 155.7, 145.6, 139.2, 135.7, 134.4, 131.9, 129.5, 128.6, 125.6, 119.7, 118.5, 117.0, 109.1, 97.8, 72.0, 71.0, 70.8, 68.0, 61.6, 20.5, 20.4, 20.4, 20.3. MS m/z 589.4 [M + Na]^+^ (calcd for C_28_H_26_N_2_O_11_, 566.15).
Compound 30c: brown solid, yield: 86.5%. ^1^H NMR (400 MHz, DMSO-d_6_) δ (ppm): 8.78 (d, J = 5.00 Hz, 1H), 8.31 (d, J = 8.52 Hz, 1H), 8.20 (d, J = 5.00 Hz, 1H), 8.15–8.08 (m, 2H), 7.21 (dd, J = 8.60, 2.32 Hz, 1H), 6.96 (d, J = 9.76 Hz, 1H), 5.70 (d, J = 7.76 Hz, 1H), 5.44–5.37 (m, 2H), 5.35–5.27 (m, 1H), 4.56 (t, J = 7.32 Hz, 1H), 4.19–4.08 (m, 2H), 2.17 (s, 3H), 2.09 (s, 3H), 2.04 (s, 3H), 1.97 (s, 3H). ^13^C NMR (100 MHz, DMSO-d_6_) δ (ppm): 170.0, 170.0, 169.5, 169.3, 158.9, 158.5, 146.0, 140.1, 139.9, 135.4, 132.0, 129.1, 128.3, 124.7, 119.2, 116.8, 114.6, 104.3, 98.0, 70.7, 70.0, 68.4, 67.3, 61.5, 20.5, 20.5, 20.4, 20.4. MS m/z 589.3 [M + Na]^+^ (calcd for C_28_H_26_N_2_O_11_, 566.15).
Compound 30d: light yellow solid, yield: 85.9%. ^1^H NMR (400 MHz, DMSO-d_6_) δ (ppm): 8.85 (d, J = 5.00 Hz, 1H), 8.44 (d, J = 8.92 Hz, 1H), 8.30 (d, J = 5.04 Hz, 1H), 8.13 (d, J = 9.80 Hz, 1H), 8.03 (d, J = 2.52 Hz, 1H), 7.42 (dd, J = 8.92, 2.52 Hz, 1H), 7.00 (d, J = 9.76 Hz, 1H), 5.62 (d, J = 7.12 Hz, 1H), 5.39 (d, J = 2.88 Hz, 1H), 5.35–5.26 (m, 2H), 4.51 (t, J = 6.40 Hz, 1H), 4.15 (d, J = 6.36 Hz, 2H), 2.17 (s, 3H), 2.10 (s, 3H), 2.00 (s, 3H), 1.97 (s, 3H). ^13^C NMR (100 MHz, DMSO-d_6_) δ (ppm): 170.0, 169.9, 169.6, 169.4, 158.7, 154.3, 145.9, 139.7, 136.0 134.5, 132.1, 129.2, 128.7, 125.5, 119.7, 117.5, 117.1, 111.5, 98.4, 70.5, 70.2, 68.4, 67.2, 61.3, 20.6, 20.5, 20.5, 20.4. MS m/z 589.4 [M + Na]^+^ (calcd for C_28_H_26_N_2_O_11_, 566.15).
Compound 30e: yellow solid, yield: 44.5%. ^1^H NMR (400 MHz, DMSO-d_6_) δ (ppm): 8.86 (d, J = 4.96 Hz, 1H), 8.46 (d, J = 8.80 Hz, 1H), 8.33 (d, J = 5.00 Hz, 1H), 8.19 (d, J = 2.32 Hz, 1H), 8.15 (dt, J = 9.80, 2.76 Hz, 1H), 7.55 (dd, J = 8.84, 2.40 Hz, 1H), 7.00 (d, J = 9.76 Hz, 1H), 5.92 (d, J = 5.48 Hz, 1H), 5.41 (d, J = 3.84 Hz, 1H), 5.34–5.23 (m, 2H), 4.99 (d, J = 9.80 Hz, 1H), 4.96 (d, J = 4.48 Hz, 1H), 4.83 (dd, J = 10.36, 3.84 Hz, 1H), 4.50–4.46 (m, 1H), 4.28 (dd, J = 12.32, 2.24 Hz, 1H), 4.17–4.11 (m, 2H), 4.00–3.97 (m, 1H), 3.95–3.90 (m, 1H), 3.73 (d, J = 8.12 Hz, 1H), 2.05 (s, 3H), 2.03 (s, 3H), 2.02 (s, 3H), 2.00 (s, 3H), 1.99 (s, 3H), 1.98 (s, 3H), 1.95 (s, 3H). ^13^C NMR (100 MHz, DMSO-d_6_) δ (ppm): 170.3, 170.3, 170.0, 169.8, 169.5, 169.4, 169.2, 158.7, 150.3, 145.8, 139.8, 138.6, 135.9, 135.6, 132.0, 129.2, 128.6, 125.8, 125.3, 122.5, 117.9, 117.6, 116.8, 72.4, 71.8, 69.6, 69.0, 67.8, 67.5, 63.7, 63.5, 61.7, 59.7, 21.3, 20.8, 20.7, 20.6, 20.4, 20.3, 20.3. MS m/z 877.1 [M + Na]^+^ (calcd for C_40_H_42_N_2_O_19_, 854.24).
3.2.11. Synthesis of Compounds 12, 13 and 31(c-e)
A solution of compound 30 (0.05 mmol) in a mixture of MeOH (2.7 mL) and H_2_O (0.5 mL) was heated to reflux with K_2_CO_3_ (40.1 mg) for 2–3 h. After removing MeOH and H_2_O under reduced pressure, the residue was subjected to silica gel column and purified using a stepwise gradient of CH_2_Cl_2_/MeOH (4:1 to 1:1), yielding pure products.
Compound 12: yellow solid, yield: 86.6%. ^1^H NMR (400 MHz, DMSO-d_6_) δ (ppm): 8.80 (d, J = 5.00 Hz, 1H), 8.31 (d, J = 8.64 Hz, 1H), 8.23 (d, J = 5.00 Hz, 1H), 8.19 (d, J = 2.24 Hz, 1H), 8.14 (d, J = 9.76 Hz, 1H), 7.32 (dd, J = 8.60, 2.32 Hz, 1H), 6.99 (d, J = 9.80 Hz, 1H), 5.46 (br s, 1H), 5.15 (br s, 2H), 5.03 (d, J = 7.20 Hz, 1H), 4.60 (br s, 1H), 3.73 (dd, J = 11.96, 2.20 Hz, 1H), 3.54 (dd, J = 12.00, 5.16 Hz, 1H), 3.30–3.26 (m, 2H), 3.26–3.18 (m, 2H). ^13^C NMR (100 MHz, DMSO-d_6_) δ (ppm): 159.8, 159.0, 146.1, 140.1, 140.1, 135.4, 132.0, 129.5, 128.4, 124.6, 118.4, 116.7, 114.4, 104.6, 101.1, 77.2, 76.5, 73.3, 69.5, 60.5. MS m/z 421.1 [M + Na]^+^ (calcd for C_20_H_18_N_2_O_7_, 398.11).
Compound 13: yellow solid, yield: 83.2%. ^1^H NMR (400 MHz, DMSO-d_6_) δ (ppm): 8.85 (d, J = 5.00 Hz, 1H), 8.41 (d, J = 8.92 Hz, 1H), 8.30 (d, J = 5.04 Hz, 1H), 8.14 (d, J = 9.76 Hz, 1H), 8.09 (d, J = 2.52 Hz, 1H), 7.45 (dd, J = 8.92, 2.60 Hz, 1H), 7.00 (d, J = 9.80 Hz, 1H), 5.02 (d, J = 7.16 Hz, 1H), 3.71 (dd, J = 12.00, 2.20 Hz, 1H), 3.54 (d, J = 5.56 Hz, 1H), 3.51 (d, J = 5.52 Hz, 1H), 3.30–3.26 (m, 2H), 3.20 (d, J = 8.84 Hz, 1H). ^13^C NMR (100 MHz, DMSO-d_6_) δ (ppm): 158.7, 155.6, 145.9, 139.6, 135.9, 133.8, 132.0, 129.6, 128.8, 125.4, 120.7, 117.6, 116.9, 110.8, 101.2, 77.3, 76.8, 73.4, 69.7, 60.7. MS m/z 421.1 [M + Na]^+^ (calcd for C_20_H_18_N_2_O_7_, 398.11).
Compound 31c: white solid, yield: 94.1%. ^1^H NMR (400 MHz, DMSO-d_6_) δ (ppm): 8.76 (d, J = 5.00 Hz, 1H), 8.24 (d, J = 8.64 Hz, 1H), 8.16 (d, J = 5.00 Hz, 1H), 8.14 (d, J = 2.36 Hz, 1H), 8.09 (d, J = 9.76 Hz, 1H), 7.28 (dd, J = 8.68, 2.32 Hz, 1H), 6.94 (d, J = 9.80 Hz, 1H), 5.00 (d, J = 7.56 Hz, 1H), 3.77 (d, J = 3.32 Hz, 1H), 3.70–3.64 (m, 2H), 3.62–3.53 (m, 2H), 3.53–3.48 (m, 1H). ^13^C NMR (100 MHz, DMSO-d_6_) δ (ppm): 159.9, 158.9, 146.0, 140.0, 135.3, 131.9, 129.4, 128.3, 124.5, 118.2, 116.6, 114.5, 104.4, 101.6, 75.6, 73.2, 70.4, 68.0, 60.2. MS m/z 421.0 [M + Na]^+^ (calcd for C_20_H_18_N_2_O_7_, 398.11).
Compound 31d: white solid, yield: 92.7%. ^1^H NMR (400 MHz, DMSO-d_6_) δ (ppm): 8.83 (d, J = 5.00 Hz, 1H), 8.38 (d, J = 8.88 Hz, 1H), 8.25 (d, J = 5.00 Hz, 1H), 8.11 (d, J = 9.76 Hz, 1H), 8.05 (d, J = 2.44 Hz, 1H), 7.44 (dd, J = 8.88, 2.48 Hz, 1H), 6.98 (d, J = 9.76 Hz, 1H), 5.31(br s, 1H), 4.99 (d, J = 7.64 Hz, 1H), 4.95 (br s, 1H), 4.74 (br s, 1H), 4.59 (br s, 1H), 3.75 (d, J = 3.28 Hz, 1H), 3.70–3.63 (m, 2H), 3.61–3.52 (m, 2H), 3.46 (dd, J = 9.60, 3.24 Hz, 1H). ^13^C NMR (100 MHz, DMSO-d_6_) δ (ppm): 158.6, 155.6, 145.8, 139.5, 135.9, 133.7, 132.0, 129.4, 128.7, 125.3, 120.0, 117.4, 116.9, 110.9, 101.8, 75.7, 73.4, 70.4, 68.2, 60.4. MS m/z 421.1 [M + Na]^+^ (calcd for C_20_H_18_N_2_O_7_, 398.11).
Compound 31e: yellow solid, yield: 75.6%. ^1^H NMR (400 MHz, DMSO-d_6_) δ (ppm): 8.85 (d, J = 5.00 Hz, 1H), 8.42 (d, J = 8.92 Hz, 1H), 8.29 (d, J = 5.00 Hz, 1H), 8.14 (d, J = 9.76 Hz, 1H), 8.09 (d, J = 2.44 Hz, 1H), 7.46 (dd, J = 8.92, 2.48 Hz, 1H), 7.00 (d, J = 9.76 Hz, 1H), 5.10 (d, J = 7.68 Hz, 1H), 5.07 (d, J = 3.76 Hz, 1H), 3.79 (d, J = 11.08 Hz, 1H), 3.70–3.58 (m, 4H), 3.54–3.42 (m, 6H), 3.17 (s, 1H), 3.09 (t, J = 9.00 Hz, 2H). ^13^C NMR (100 MHz, DMSO-d_6_) δ (ppm): 158.7, 155.4, 150.1, 145.9, 135.9, 133.9, 132.0, 129.5, 128.7, 125.4, 120.0, 117.5, 116.9, 110.8, 100.9, 100.8, 79.2, 76.4, 75.4, 73.7, 73.3, 72.9, 72.5, 69.9, 60.9, 60.3. MS m/z 559.2 [M − H]^−^ (calcd for C_26_H_28_N_2_O_12_, 560.16).
3.3. Microbroth Dilution Assay
The bioassay studies employed four microbial human pathogen strains: Staphylococcus aureus (ATCC 43300), Escherichia coli (ATCC 25922), Candida albicans (ATCC 10231) and Cryptococcus neoformans (ATCC 66031). Bacterial pathogens S. aureus and E. coli were maintained in Tryptic Soy media (TSA, TSB; BD Biosciences, San Jose, CA, USA) at 37 °C. Fungal microorganisms C. albicans and C. neoformans were propagated in yeast malt (YM; BD Biosciences) and Sabouraud dextrose media (SDA; BD Biosciences), respectively, at 30 °C. All liquid cultures were incubated with constant agitation at 200 rpm using an orbital shaker.
Antimicrobial activity evaluation was performed through broth microdilution method [34]. Test compounds were initially solubilized in DMSO to achieve a 20 mg/mL stock solution, which was subsequently stored at 4 °C until experimental use. Microbial suspensions were prepared from overnight cultures, adjusted to an optical density (OD_600_) ranging from 0.03 to 0.06, representing the exponential growth phase. These suspensions were then diluted to 1:10 in appropriate broth media (except for C. neoformans, which was used undiluted) and aliquoted into 96-well microplates (195 μL/well). Following incubation periods of 24 h for bacterial strains and 48 h for fungal strains, optical density measurements were recorded at 600 nm. The percentage inhibition was determined by subtracting the blank well absorbance from sample readings, normalizing against the average absorbance of DMSO control wells and multiplying by 100.
4. Conclusions
In summary, the efficient synthesis of canthin-6-one alkaloids 4, 5, 7 and 12-13 and 8 novel derivatives was achieved for the first time. The synthetic process involved key Pd-catalyzed Suzuki–Miyaura coupling, Cu-catalyzed amidation and Koenig–Knorr glycosylation reactions, using 16 as the starting material. Antimicrobial activity was evaluated, revealing that canthin-6-one alkaloids 1-4 and 7 exhibited antimicrobial properties against C. albicans, C. neoformans and S. aureus comparable to those of the positive control drugs at 50 µg/mL. The antimicrobial activity of alkaloids 2 and 4-5 against these four microbial human pathogens and canthin-6-one alkaloids 6-7 against C. albicans, C. neoformans and E. coli is reported here for the first time. However, none of the derivatives demonstrated antimicrobial activity, and all synthesized compounds were ineffective against E. coli at 50 µg/mL. It can be concluded that the substituents on the A-ring of canthin-6-one play a crucial role in its antimicrobial properties. This study lays the foundation for the further development of antimicrobial agents.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Dai W.Q. He J.F. Ye F. Xu J. Liu W.Y. Feng F. Qu W. Design, Synthesis and in Vitro Biological Evaluation of Novel 4-Methoxy-5-Hydroxycanthin-6-One Derivatives as Potential Anti-Tumor Agents Nat. Prod. Res.202034228922943060069910.1080/14786419.2018.1536708 · doi ↗ · pubmed ↗
- 2Qiu S. Sun H. Zhang A.H. Xu H.Y. Yan G.L. Han Y. Wang X.J. Natural Alkaloids: Basic Aspects, Biological Roles, and Future Perspectives Chin. J. Nat. Med.20141240140610.1016/S 1875-5364(14)60063-724969519 · doi ↗ · pubmed ↗
- 3Ng Y.P. Or T.C.T. Ip N.Y. Plant Alkaloids as Drug Leads for Alzheimer’s Disease Neurochem. Int.2015892602702622090110.1016/j.neuint.2015.07.018 · doi ↗ · pubmed ↗
- 4Thawabteh A. Juma S. Bader M. Karaman D. Scrano L. Bufo S.A. Karaman R. The biological activity of natural alkaloids against herbivores, cancerous cells and pathogens Toxins 20191165610.3390/toxins 1111065631717922 PMC 6891610 · doi ↗ · pubmed ↗
- 5Haynes H.F. Nelson E.R. Price J.R. Alkaloids of the Australian Rutaceae—Pentaceras-Australis Hook F. I. Isolation of the Alkaloids and Identification of Canthin-6-One Aust. J. Sci. Res. Ser. A-Phys. Sci.1952538740010.1071/CH 9520387 · doi ↗
- 6Farouil L. Sylvestre M. Fournet A. Cebrián-Torrejón G. Review on Canthin-6-One Alkaloids: Distribution, Chemical Aspects and Biological Activities Eur. J. Med. Chem. Rep.202252110.1016/j.ejmcr.2022.100049 · doi ↗
- 7Brahmbhatt K.G. Ahmed N. Sabde S. Mitra D. Singh I.P. Bhutani K.K. Synthesis and Evaluation of Β-Carboline Derivatives as Inhibitors of Human Immunodeficiency Virus Bioorg. Med. Chem. Lett.2010204416441910.1016/j.bmcl.2010.06.05220598537 · doi ↗ · pubmed ↗
- 8Zhang Y. Liu Y.B. Li Y. Ma S.G. Li L. Qu J. Zhang D. Chen X.G. Jiang J.D. Yu S.S. Sesquiterpenes and Alkaloids from the Roots of Alangium Chinense J. Nat. Prod.2013761058106310.1021/np 400074723734721 · doi ↗ · pubmed ↗