Current pattern of antibiotic resistance and molecular characterization of virulence genes in Klebsiella pneumoniae obtained from urinary tract infection (UTIs) patients, Peshawar
Zeeshan Khan, Qaisar Ali, Sadiq Azam, Ibrar Khan, Jamila Javed, Noor Rehman, Mesaik M. Ahmed, Jalal Uddin, Ajmal Khan, Ahmed Al-Harrasi, Yung-Fu Chang, Yung-Fu Chang, Yung-Fu Chang

TL;DR
This study examines antibiotic resistance and virulence genes in Klebsiella pneumoniae from UTI patients in Peshawar, highlighting the growing threat of drug-resistant infections.
Contribution
The study provides a detailed molecular characterization of virulence genes and antibiotic resistance patterns in clinical isolates of K. pneumoniae from UTIs in a specific hospital setting.
Findings
K. pneumoniae showed high resistance to trimethoprim/Sulfamethoxazole and Colistin, with Tigecycline and Cefepime being the most effective antibiotics.
The fimH virulence gene was most prevalent, and mutations were observed in fimH and papEF, but not in sat, afa, or vat.
Significant associations between bacterial types were found, supporting the null hypothesis with p ≤ 0.05.
Abstract
The current study investigates the prevalence of virulence genes obtained from clinical isolates of multidrug-resistant (MDR) Klebsiella pneumoniae at Khyber Teaching Hospital Peshawar, from October 2021 to January 2023. Upon proper consent, clinical samples of suspected UTIs patients were collected and inoculated on the nutrients agar media, McConkey agar media, and Cysteine Lysine Electrolyte Deficient (CLED) agar media followed by incubation at 37°C for 24 hrs. The phenotypic and genotypic identification were employed for the bacterial isolates. The phenotypic identification includes gram staining followed by the Analytical Profile Index (API 20E). A total of 215 (3.85%) positive isolates were found with the highest prevalence observed among the female patients (4.35%) followed by male (3.26%). The highest prevalence, constituting 52.55% (n = 113), was detected in the age group of…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Fig 1
Fig 1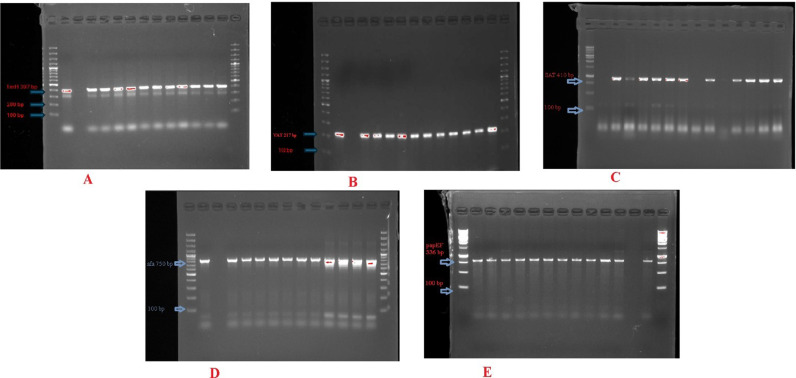 Fig 2
Fig 2- —http://dx.doi.org/10.13039/501100007446King Khalid University
- —The Oman Rese
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAntibiotic Resistance in Bacteria · Urinary Tract Infections Management · Bacterial Identification and Susceptibility Testing
Introduction
Klebsiella pneumoniae is a Gram-negative, non-motile, facultative anaerobic bacterium in rod-shaped forms, varying in size from 1–2µm. It is commonly found in different strains as commensal, with some strains being pathogenic to humans [1]. Typically found in the human intestine, K. pneumoniae can cause various infections, including bacteremia, suppurative infections, soft tissue infections, osteomyelitis, or meningitis, and usually immunocompromised patients. Sometimes it colonizes human mucosal surfaces in the oropharynx and gastrointestinal (GI) tract [2]. K. pneumoniae is a member of the ESKAPE (Enterococcus spp, staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp) group and is considered the second-most uropathogenic bacterium after E. coli [3]. This bacterial species is a significant challenge due to its MDR, as it is highly reported for producing β-lactamases. Such resistance leads to limited treatment options. However, the variation in the pathogenicity of the K. pneumoniae strains can be attributed to the presence or expression of the virulence factors [4]. The enhancement of K. pneumoniaes’s pathogenicity is attributed to the presence of the MDR virulence Mega plasmid. Well-characterized virulence factors in K. pneumoniae include siderophores, lipopolysaccharides, and fimbriae [5]. These essential virulence factors are crucial in the initial stages of infection [6]. These structural components are critical in adherence, invasion, and development of infections [7]. In recent studies, several virulence factors, including porins, outer membrane proteins, iron transport systems, efflux pumps, and genes involved in allantoin metabolism, have been thoroughly characterized. Surface anchored proteins (SAT) are involved in the adhesion of bacterium to host cells [8]. Once attached to the host cell, Sat triggers the activation of signaling pathways that lead to the uptake of K. pneumoniae by the host cell. Type 1 fimbrial adhesin is found on the tip of fimbriae, which are hair-like structures that extend from the surface of K. pneumoniae [9]. The fimH is responsible for the binding of K. pneumoniae to specific carbohydrate molecules on the surface of host cells particularly in the urinary and respiratory tracts [9]. This interaction also ends in bacterial uptake. The absence of either Sat or FimH significantly weakens K. pneumoniae virulence. Additionally, a multitude of factors, including the capsule, lipopolysaccharide, siderophores, and efflux pumps, contribute to the pathogen’s virulence [10]. The presence of virulence factors and drug resistance mechanisms enhances the pathogen’s capability to establish and persist in the colonized form. Therefore, considering the pathogenicity of K. pneumoniae strains, molecular characterization of virulence genes in both community and hospital settings is necessary for the development of potential antibiotic therapies. Different studies have been reported on the epidemiology of resistant genes, but limited research has addressed the genomics and pathogenicity of different strains [11]. Therefore, the current research work was conducted to assess antibiotic susceptibility testing and molecular characterization of virulence genes in clinical isolates of K. pneumoniae obtained from UTIs patients in KTH, Peshawar.
Methodology
Isolation and identification of clinical samples
The current research study was conducted in the pathology laboratory at KTH, Peshawar, and the Molecular and Genomics laboratory, at the Center of Biotechnology and Microbiology, University of Peshawar from October 2021 to January 2023. Upon proper consent, the patient’s medical history, along with various parameters such as gender and age distribution. The study was approved by Khyber Medical College Peshawar Institutional Research and Ethical Board (IREB) Pakistan (No.124/DME/KMC).
Urine samples were collected from the UTIs patients and were inoculated on the nutrients agar media, McConkey agar media, and CLED agar media. The growth of bacterial samples was observed after incubation at 37°C for 24 hrs. Subsequently, both Phenotypic and genotypic identification were employed to identify the bacterial isolates. The phenotypic identification includes gram staining followed by the API 20E.
Extraction of genomic DNA
Genomic DNA was extracted from the 24 h old culture of confirmed bacterial isolates using a Thermo Scientific DNA Purification Kit. A 1% agarose gel was prepared in 1X Tris Acetate EDTA (TAE) buffer, and gel electrophoresis was conducted to confirm the presence of DNA. Gel Doc^TM^ Bio-Rad Molecular imager® was utilized to visualize the bands.
Molecular identification of bacterial isolates
Genotypic identification via a specific primer of WBBZ (567bp) as shown in Table 2, under optimized conditions was performed for confirmation of the bacterial isolates. The specific gene was amplified, followed by electrophoresis on a 2% agarose Gel. The resulting bands were visualized by using the Gel Doc^TM^ Bio-Rad Molecular imager®.
Table 2: Specific primers sequence used for molecular identification and virulence genes.
Antibiotic susceptibility testing
Using the Kirby Bauer disc diffusion method, the antibiotic susceptibility pattern of bacterial isolates was determined against various groups of antibiotics following the guidelines of the Clinical and Laboratory Institutes (CLSI-2022). A standardized bacterial suspension equivalent to 0.5 McFarland turbidity was prepared from the isolate. The suspension was evenly spread across the surface of a sterile Muller Hinton agar (MHA) plate using a sterile cotton swab to create a uniform lawn of bacteria. The swab was passed in three directions to ensure consistent coverage, with a final sweep along the edge of the plate. The agar plate was allowed to dry for a few minutes at room temperature to ensure proper adherence to the bacterial inoculum. Next, selected antibiotic discs were carefully placed on the agar surface using sterile forceps, ensuring even spacing to avoid overlapping zones of inhibition. The plates were then incubated for 24 h at 37°C in an inverted position to prevent condensation from affecting the diffusion of antibiotics. After incubation, the zones of inhibition around each disc were measured in millimeters. The results were interpreted according to CLSI guidelines as intermediate (I), resistant (R), or sensitive (S) as shown in Table 1.
Table 1: Selected specific antibiotics and their concentration used in the current research study.
Molecular characterization of the virulence genes
Specific PCR primers for the virulence genes as shown in Table 2, under optimized conditions, were employed in the investigation of all bacterial isolates. A total PCR reaction volume of 27 µl was prepared by combining 12.5 µl of 2x Thermo scientific master mix, 1 µl of forward and reverse primer, 2 µl of DNA template, and the remaining volume was filled with PCR grade water. The amplified products were then run on 2% agarose gel and visualized using the Del Doc® system.
Minimum inhibitory concentration (MIC)
The effectiveness of antibiotics is determined by their MIC values by using E-strips as shown in Table 3. The clinical isolates were inoculated on MHA media and strips were employed followed by incubation at 37°C for 24 h.
Table 3: E-Strip used for the MIC determination.
Mutational analysis of PCR products
The amplified DNA was sent to the Genomic and Sequencing Laboratory of Khyber Medical University, Peshawar for sequencing by using the Sanger Sequencing method. The sequencing data were analyzed by different bioinformatics tools such as Bio-edit sequence alignment editor, and CLUSTLW. The consensus sequences were generated for each gene and checked through the NCBI Basic Local Alignment Search Tool (BLAST) to determine the local similarity between them.
Statistical analysis
An analysis using SPSS version 20 utilized chi-square to determine the association between the expected value and the observed value, revealing a significance level of p ≤ 0.05. This analysis was conducted with a sample size (n) of 150, employing degrees of freedom calculated as n-1.
Results
Isolation and identification of Klebsiella pneumoniae
A total of 5,580 clinical isolates were analyzed, of which 215 were detected positive for K. pneumoniae, isolated in KTH, Peshawar from Urine samples. The highest prevalence, constituting 52.55% (n = 113), was detected in the age group of 21–40 years, followed by 31.62% (n = 68) in the 41-60 years age group. Additionally, 10.23% (n = 22), 3.25% (n = 7), and 2.32% (n = 5) of cases were identified in the age groups of 01–10 years, 11-20 years, and above 60 years, respectively. Among the total positive samples, 44.65% (n = 96) were collected from the Outpatient department (OPD), while inpatient department (IPD) cases contributed 55.35% (n = 119) as shown in Table 4. For the precise and accurate identification of K. pneumoniaee clinical isolates, the API kit was employed as shown in Fig 1 and S3 Fig. The API system includes a series of biochemical tests designed to exploit the metabolic activities of bacteria. Positive results were observed for catalase and urease activity, demonstrating the organism’s ability to hydrolyze urea and produce ammonia. Additionally, K. pneumoniaee exhibited positive fermentation of glucose and lactose, with acid detected as the product. Other biochemical tests, including indole production, phenylalanine deaminase activity, sucrose fermentation, and citrate utilization, also yielded positive results, further confirming the identity of the isolate.
Table 4: Different parameters of the clinical isolates obtained from UTIs patients.
Analytical profile index result for the identification of Klebsiella pneumoniae.
Antibiotic susceptibility testing
The antibiotic-sensitivity pattern of K. pneumoniae revealed the resistance-sensitive percentage values in the Table 2. Especially, SXT stands out with the highest resistance rate, reaching 93%. Following closely is CO, exhibiting a resistance rate of 79.07%. Especially, SXT stands out with the highest resistance rate, reaching 93%. Following closely is CO, exhibiting a resistance rate of 79.07%. The TGC emerges as the most effective antibiotic, demonstrating a sensitivity rate of 90%. Similarly, FEP shows a high sensitivity level, also standing at 90%. The overall values are presented in Table 5.
Table 5: Antibiotic susceptibility pattern of K. pneumoniae against selected antibiotics.
Minimum inhibitory concentration (MIC)
The potency of antibiotics can be determined by their MIC valves, with MIC valves signifying greater effectiveness and higher MIC valves indicating diminished potency (S1, S2, S5 and S6 Figs). The K. pneumoniae causing UTIs obtained from the KTH, Peshawar revealed more resistance in the case of CTX, MEM, CN, AK, DO, CIP, and SXT as shown in Table 6.
Table 6: MIC of the selected antibiotic used against K. pneumoniae.
Comprehensive molecular characterization of virulence genes in Klebsiella pneumoniae
A detailed analysis of the virulence genes among the total isolates of K. pneumoniae reveals the highest prevalence of fimH (80%) followed by SAT (65%), papEF (49%), and afa (29%). The lowest prevalence of VAT (16%) was recorded as shown in Table 7 and Fig 2 and S4 and S7 Figs.
Table 7: Distribution of virulence genes detected in K. pneumoniae.
(A) L2-L7 showing PCR products of FimH (397 bp); (B) L2-L9 representing PCR products of VAT (217 bp); (C) DNA ladder of 100 bp in L1 and L8, with L2-L7 showing PCR products of SAT (410 bp); (D) L1 and L10 illustrating a 100 bp DNA ladder, with L2-L9 representing PCR products of the afa gene (750 bp), (E) DNA ladder of 100 bp in L1 and L9, with L2-L8 showing PCR products of papEF (336 bp).
Combination of different virulence genes in K. pneumoniae
The distribution of virulence gene combinations is briefly presented, detailing the number of genes, occurrence frequency, and percentage for each combination. The most prevalent combination is Sat/fimH/papEF which is observed in 24 isolates accounting for 11.01% of the total occurrences. Following closely is Sat/fimH, with two genes occurring 21 times, constituting 9.5% of total isolates. Other distinguished combinations include fimH/papEF (2 genes, 15 occurrences, 6.9%), Sat/fimH/afa (3 genes, 11 occurrences, 5.19%), Sat/fimH/afa/vat (4 genes, 10 occurrences, 4.5%), Sat/afa/vat (3 genes, 9 occurrences, 4.34%), Sat/fimH/papEF/vat (5 genes, 6 occurrences, 2.9%), Sat/fimH/papEF/afa (4 genes, 7 occurrences, 3.32%), and Sat/fimH/papEF/vat/afa (5 genes, 6 occurrences, 2.7%) as shown in Table 8.
Table 8: Combination of the virulence genes and their frequency detected in K. pneumoniae.
Distribution and prevalence of virulence gene combinations in clinical isolates
The analysis reveals a significant statistical correlation between antibiotic resistance phenotypes and the presence of virulence genes. The presence of different virotypes showed a statistically insignificant association (P value > 0.05) with antimicrobial resistance phenotypes. An inverse relationship was observed across all studied genes, virotypes, and phenotypic antibiotic resistance. The odds ratio indicated a negative correlation, with values less than 1. The various combinations of virulence genes and their corresponding association data are presented in Table 9.
Table 9: Combination of the virulence gene association with antibiotic association frequency.
Statistical analysis
The Chi-square test indicated a significant association between the type of bacteria, supporting our null hypothesis with a significance level of p ≤ 0.05. The one-way ANOVA test demonstrated a noteworthy relationship between the dependent and independent variables.
Discussion
Multidrug resistance K. pneumoniae is one of the leading causes of life-threatening among several infections globally. Ghafourian et al. reported that the extensive utilization of antimicrobial agents has contributed to the increase in the prevalence of MDR K. pneumoniae [15]. Ciccozzi et al. reported in the study that the rising prevalence of MDR in K. pneumoniae strains is a significant and determined public health risk, contributing to elevated levels of illness and death globally [16]. Manjula et al. reported that Gram-negative bacteria have developed mechanisms to resist existing antibiotics, highlighting the challenge in treating bacterial infections. Similarly, in the current research study, the highest prevalence rate of 89% MDR K. pneumoniae was identified, consistent with the 90.2% prevalence by Manjula et al. in the literature. In the reported study most of the strains showed resistance to various antibiotics including cephalosporins, penicillin, fluoroquinolones, sulfonamides, and aminoglycosides [17]. The antibiotic susceptibility profile of K. pneumoniae showed significant resistance to Amoxicillin/Clavulanate (87%), trimethoprim/Sulfamethoxazole (93%), and Colistin (79.07%). The reported study reveals that MDR K. pneumoniae is responsible for hospital-acquired infection and was found highly resistant toward tetracycline (95.2%), ciprofloxacin, and gentamycin (76.5% each), sulphathiazole (66.7%), nalidixic acid (61.9%) and norfloxacin (42.9%) [18]. Similarly, Indrajitha et al. (2021) reported a study regarding the drug resistance of K. pneumoniae which highlighted the 38% resistance to imipenem and 31% to meropenem respectively. The highest prevalence rate was detected in female patients (n = 131, 4.3%), followed by male patients (n = 84, 3.26%) in clinical isolates of K. pneumoniae in the current research study as supported by the reported study [19]. The prevalence of K. pneumoniae infection is increasing in Pakistan which is directly aligned with an increase in antibiotic resistance. This is consistent with the findings of Martin et al. (2016), who reported a 23% infection rate, and Zhang et al. (2018), who reported a 73.9% infection rate [20,21]. In the present study, the high prevalence rate of virulence genes such as fimH (80%), SAT (65%), papEF (49%), afa (29%), and VAT (16%) in the MDR K. pneumoniae were observed. The strains obtained from patients with UTIs were investigated to determine their high potential rate of pathogenicity. A similar study of the virulence genes was reported in hospital-acquired infection caused by K. pneumoniae strains in China, the UK, France, and Brazil [22–25]. According to the reported study, Type-I fimbriae is the most common and frequent adhesive factor in the strains of K. pneumoniae. However, the presence of this gene has been associated with increased susceptibility to UTIs. This type 1 fimbrial adhesion has been identified as a mediating factor in the binding of K. pneumoniae strains to the mucous tissue layer of respiratory and Urinary tracts [26]. Wasfi et al. (2016) reported that most of the MDR strains of K. pneumoniae express type-I fimbrial adhesions. Similarly in the current research study, the prevalence of FimH (80%), papEF (49%), and afa (29%), adhesive gene was identified [26]. Literature has reported that focused on the characterization of virulence factors in K. pneumoniae isolates performed statistical tests to evaluate the prevalence of fimH, which is crucial for adhesion and biofilm formation. The study utilized chi-square tests to analyze the correlation between the presence of fimH and the severity of UTIs, indicating a statistically significant association (p ≤ 0.05) between fimH expression and increased virulence [27]. Another research paper assessed the prevalence of various virulence genes, including afa, vat, sat, and papEF, among clinical isolates of K. pneumoniae. One-way ANOVA was employed to compare the mean expression levels of these genes in multidrug-resistant (MDR) and non-MDR strains. The results showed significant differences (p ≤ 0.05) in the expression of vat and papEf among different resistance profiles, revealing their potential role in the pathogenicity of MDR strain [28]. Similarly, in this current study, the virulence gene’s association with antibiotic resistance was determined. The analyzed study statistically correlates antibiotic resistance phenotypes and virulence genes. The presence of various virotypes demonstrated a varied statistical connection (P value > 0.05) with antimicrobial resistance phenotypes. Overall, there was a noted inverse association observed among all studied genes, virotypes, and phenotypic antibiotic resistance. The odds ratio revealed a negative correlation, representing a value of less than 1. The finding and mutation in the virulence genes may offer a molecular explanation of antibiotic resistance observed in the isolates of the conducted study.
Conclusion
The study highlights the critical global threat of antibiotic resistance, particularly in K. pneumoniae. The observed resistance in Gram-negative bacteria, remarkable to key antibiotics, signals a pressing need for alternative treatment strategies. The prevalence of MDR K. pneumoniae in hospital-acquired infections, especially among female patients, mirrors an alarming trend in Pakistan. The investigation into virulence gene expression provides valuable awareness of the molecular basis of antibiotic resistance. Urgent and concerted efforts are needed to address this growing challenge and develop effective therapeutic approaches against MDR bacterial infections.
Supporting information
S1 FigBacterial growth on MacConkey agar media.(JPG)
S2 FigGram staining of K. Pneumoniae.(JPG)
S3 FigAnalytical Profile Index for the identification of K. pneumoniae.(JPG)
S4 FigGel image of Wbbz (567 bp) for Molecular identification of K. pneumoniae: L1,L10: DNA ladder 100 bp, L2-L9: PCR product of Wbbz Gene.(JPG)
S5 FigRepresentative image of Antibiogram of K. pneumoniae.(JPG)
S6 FigRepresentative image of determination of MIC of K. pneumoniae determined by E-strip.(JPG)
S7 FigOriginal uncropped images of gel of Fig 2 of the main text.(DOCX)
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Rønning TG, Aas CG, Støen R, Bergh K, Afset JE, Holte MS, et al. Investigation of an outbreak caused by antibiotic-susceptible Klebsiella oxytoca in a neonatal intensive care unit in Norway. Acta Paediatr. 2019;108(1):76–82. doi: 10.1111/apa.14584 30238492 · doi ↗ · pubmed ↗
- 2Tsereteli M, Sidamonidze K, Tsereteli D, Malania L, Vashakidze E. Epidemiology of carbapenem-resistant Klebsiella pneumoniae in intensive care units of multiprofile hospitals in Tbilisi, Georgia. Georgian Med News. 2018;280(281):164–8.30204118 · pubmed ↗
- 3Mulani MS, Kamble EE, Kumkar SN, Tawre MS, Pardesi KR. Emerging strategies to combat ESKAPE pathogens in the era of antimicrobial resistance: a review. Front Microbiol. 2019;10:539.30988669 10.3389/fmicb.2019.00539 PMC 6452778 · doi ↗ · pubmed ↗
- 4Martin RM, Cao J, Wu W, Zhao L, Manthei DM, Pirani A, et al. Identification of pathogenicity-associated loci in klebsiella pneumoniae from hospitalized patients. Msystems. 2018;3(3):e 00015–18. doi: 10.1128/m Systems.00015-18 29963640 PMC 6020474 · doi ↗ · pubmed ↗
- 5Paczosa MK, Mecsas J. Klebsiella pneumoniae: Going on the offense with a strong defense. Microbiol Mol Biol Rev. 2016;80(3):629–61. doi: 10.1128/MMBR.00078-15 27307579 PMC 4981674 · doi ↗ · pubmed ↗
- 6Huynh DTN, Kim A-Y, Kim Y-R. Identification of pathogenic factors in klebsiella pneumoniae using impedimetric sensor equipped with biomimetic surfaces. Sensors (Basel). 2017;17(6):1406. doi: 10.3390/s 17061406 28617330 PMC 5492845 · doi ↗ · pubmed ↗
- 7Bien J, Sokolova O, Bozko P. Role of uropathogenic escherichia coli virulence factors in development of urinary tract infection and kidney damage. Int J Nephrol. 2012;2012:681473. doi: 10.1155/2012/681473 22506110 PMC 3312279 · doi ↗ · pubmed ↗
- 8Martin RM, Bachman MA. Colonization, infection, and the accessory genome of klebsiella pneumoniae. Front Cell Infect Microbiol. 2018;8:4. doi: 10.3389/fcimb.2018.00004 29404282 PMC 5786545 · doi ↗ · pubmed ↗