From benign appearance to malignant truth: a case report of mesenteric dedifferentiated liposarcoma with inflammatory myofibroblastic tumor-like features
Simon Schallenberg, Miriam Schulte, Mihnea P. Dragomir, Armin Jarosch, Wolfgang Hartmann, Eva Wardelmann

TL;DR
A rare case of aggressive mesenteric cancer initially mistaken for a benign tumor is reported, emphasizing the need for molecular testing.
Contribution
The paper presents a novel case of DDLPS with IMT-like features, highlighting diagnostic challenges and the role of molecular analysis.
Findings
The tumor showed morphological similarities to IMT but had distinct molecular features of DDLPS.
MDM2 gene amplification was detected, confirming the aggressive nature of the tumor.
The case underscores the importance of molecular pathology in distinguishing aggressive sarcomas from benign tumors.
Abstract
Dedifferentiated liposarcoma (DDLPS) with inflammatory myofibroblastic tumor (IMT)-like features is a rare and diagnostically challenging variant of soft tissue sarcoma. We report the case of a 74-year-old man who presented with a mesenteric mass in 2022 and recurrent tumors in 2024. Tissue from both primary and recurrent tumors were submitted to our reference center for pathological reevaluation, with a suspicion of IMT being suspected. Although the tumors exhibited morphological characteristics consistent with those observed in IMT, they displayed distinctive histological, immunohistochemical and molecular features suggestive of DDLPS with IMT-like features, including amplification of the MDM2 gene. This report highlights the morphological spectrum of DDLPS, the diagnostic role of molecular pathology, and the importance of differentiating this aggressive neoplasm from benign entities…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
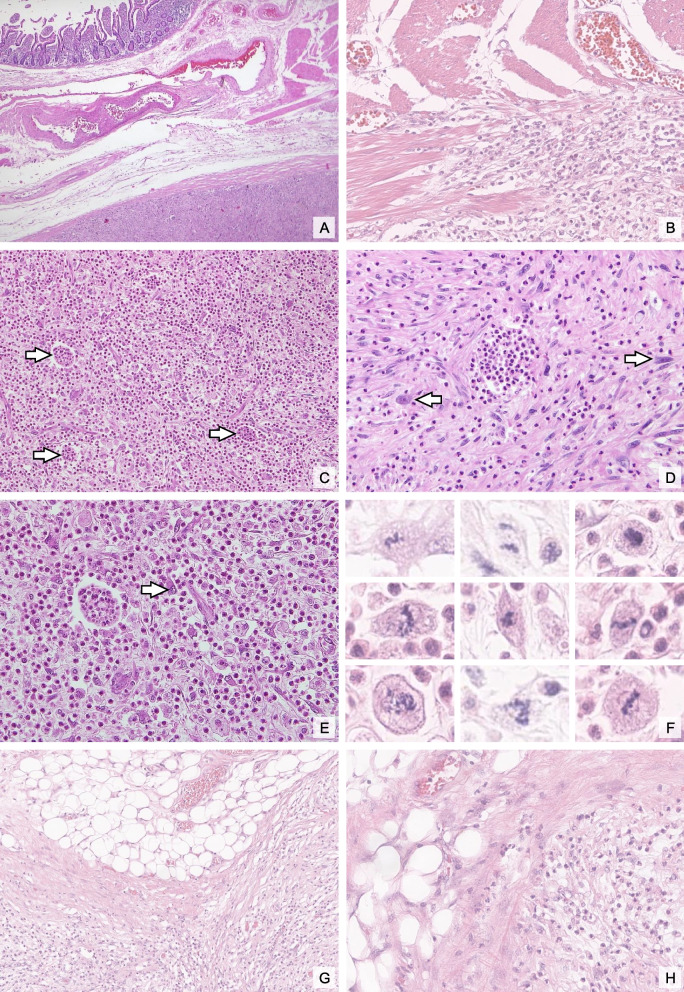 Figure 1
Figure 1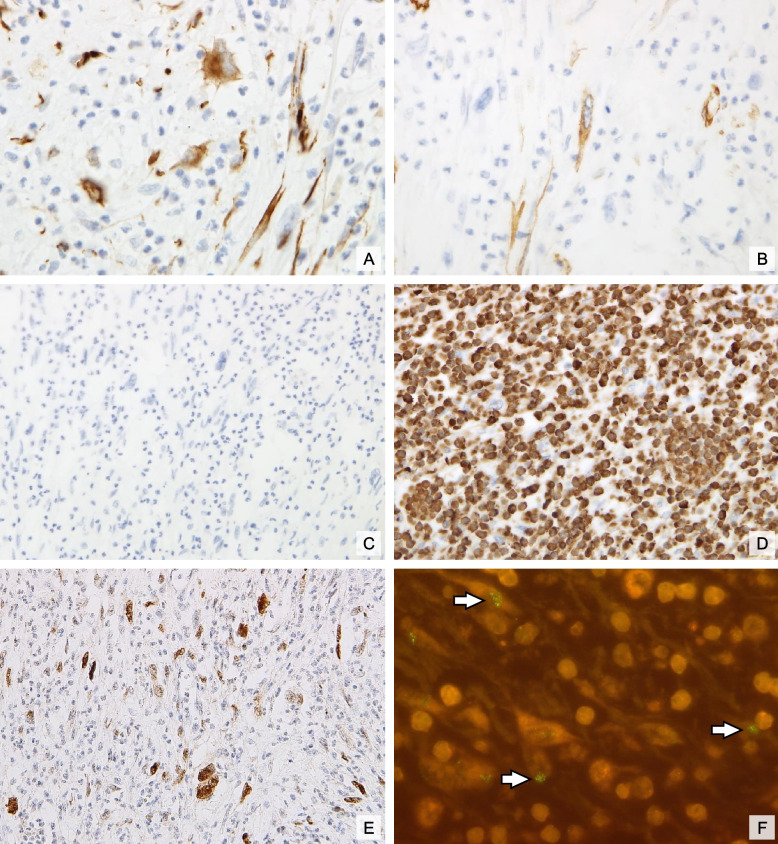 Figure 2
Figure 2- —Charité - Universitätsmedizin Berlin (3093)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsIgG4-Related and Inflammatory Diseases · Soft tissue tumor case studies · Sarcoma Diagnosis and Treatment
Introduction
Dedifferentiated liposarcoma (DDLPS) is an atypical lipomatous tumor/well-differentiated liposarcoma (ALT/WDLPS) that progresses, either in the primary or in a recurrence, to a non-lipogenic sarcoma of variable histological grade. It represents approximately 10–20% of all liposarcomas [1–3]. It has been established that approximately 90% of cases arise de novo, while 10% develop from recurrences [1–3]. DDLPS is most often found in the retroperitoneum but can also occur in the spermatic cord and more rarely in head and neck, mediastinum, trunk or mesentery [1, 4–10].
The World Health Organization (WHO) classification of soft tissue tumors delineates five main subtypes of liposarcoma: well-differentiated, dedifferentiated, myxoid, pleomorphic, and myxoid pleomorphic liposarcomas. The histological hallmark of DDLPS is the transition from the WDLPS component to a higher-grade, non-lipogenic sarcomatous component [1, 2]. Morphologically, the dedifferentiated areas may exhibit a broad histological spectrum, most frequently resembling undifferentiated pleomorphic sarcoma or leiomyosarcoma and, in rare cases, inflammatory myofibroblastic tumor (IMT)-like features [11–16].
IMT is a low-grade spindle cell neoplasm typically associated with anaplastic lymphoma kinase (ALK) and more rarely with ROS oncoprotein (ROS1) or neurotrophic tropomyosin kinase receptor (NTRK) rearrangements and a less aggressive clinical course compared to DDLPS [17–19]. IMT-like DDLPS variants, however, pose a significant diagnostic challenge due to overlapping histological features, such as myxoid, cellular, and hypocellular fibrous stroma, chronic inflammatory infiltrates as well as areas closely resembling fibromatosis or nodular fasciitis [11, 12]. Misdiagnosis of IMT-like DDLPS as a benign entity such as IMT, desmoid-type fibromatosis, and reactive myofibroblastic lesions can lead to suboptimal treatment strategies [11, 12].
Molecular pathology plays a pivotal role in resolving these diagnostic dilemmas. The amplification of MDM2, by definition detected in DDLPS, serves as a hallmark molecular signature [20]. Conversely, the absence of ALK, ROS1 or NTRK rearrangements and of CTNNB1 mutations help to exclude IMT and desmoid fibromatosis, respectively.
In this report, we present the case of a 74-year-old male with a rare mesenteric DDLPS exhibiting IMT-like features, diagnosed initially in 2022 and presenting with recurrent tumors in 2024. This case underscores the importance of integrating histopathological, immunohistochemical, and molecular findings to navigate the complex differential diagnosis of mesenchymal tumors. Additionally, we aim to provide a review of the literature, highlighting diagnostic and therapeutic implications for this rare variant of DDLPS.
Case description
A 74-year-old male patient was admitted in 2022 with a primary mesenteric mass. In 2024, three additional subserosal tumors were identified in the mesentery, with the largest measuring 5 cm. Subsequently, all tumors were excised via surgical resection. Tissues from all tumors were submitted to our reference center for pathological reevaluation, with an external diagnosis of IMT being suspected.
Histological examination and molecular pathology
The histological examination of the primary tumor revealed the presence of a centripetal infiltration of the intestinal wall, extending to the tunica muscularis propria (Fig. 1A + B). At low magnification, both primary and recurrent tumors displayed a myxoid and loosened stroma, interspersed with dense inflammatory infiltrates predominantly composed of eosinophils and neutrophils (Fig. 1C). These inflammatory cells formed microabscesses in several regions of the tumor, contributing to its distinctive appearance. At higher magnification, single bizarre tumor cells were evident, characterized by pronounced pleomorphism and hyperchromatic nuclei, indicative of malignancy (Figs. 1D and E). A marked increase in mitotic activity was observed, with up to 7 mitotic figures per 10 HPF (Fig. 1F). No necrosis was identified. The tissue surrounding the tumor consisted of morphologically unremarkable adipose tissue. No adjacent atypical lipogenic tumor component was present; however, MDM2 FISH was not performed on that component for clarification (Fig. 1G + H).Fig. 1. Histomorphological characteristics: Intestinal wall cross-section with the enteral mucosa visible in the top left and DDLPS with infiltration of the tunica muscularis propria visible at the bottom right (A). Higher magnification highlighting tumor cell infiltration of the tunica muscularis propria (B). The lower magnification shows pronounced inflammatory infiltrates, characterized by a predominance of eosinophils and neutrophils and the formation of microabscesses (white arrows; C). In the tumor areas with lower inflammatory cell density, the bizarre tumor cells with pleomorphic nuclei (white arrows) are clearly visible (D), while in tumor areas with pronounced inflammatory infiltrates (E), even in the higher magnification, the tumor cells are not clearly visible (white arrow). There was a significant increase in the number of mitoses (F). No adjacent atypical lipogenic tumor component was found (G+H); hematoxylin-eosin, original magnifications x12.5(A), x40 (C, G) and x200 (B, D, E, H) and 400x (F)
Immunohistochemically, individual tumor cells showed cytoplasmic expression of desmin and smooth muscle actin (SMA), indicating a partial myogenic differentiation (Figs. 2A and B). The tumor cells were negative for pan-Trk, ROS1, and anaplastic lymphoma kinase (ALK; Fig. 2C). Heterogeneous expression of p53 was noted, reflecting TP53 wildtype phenotype. The proliferation index, determined by Ki-67 staining, reached an average value of 20%, illustrating the high proliferative activity of the tumor cells. Immunohistochemical staining revealed strong nuclear expression of MDM2 (Fig. 2E).Fig. 2. Immunohistochemical and molecular features: Immunohistochemical reactivity for desmin (A) and smooth muscle actin (B) confirms myogenic differentiation, while the ALK staining was negative (C). CD15 marks the pronounced granulocytic infiltrate (D). Nuclear positivity for MDM2 and MDM2 amplification via FISH (MDM2 clusters are indicated by white arrows) confirms the diagnosis of DDLPS (E, F); original magnifications × 400 (A,B), × 200 (C-E) and × 600 (F)
Further confirmation through fluorescence in situ hybridization (FISH) showed high-level amplification of the MDM2 gene, evident as clustered signals (Fig. 2F). This amplification of MDM2, combined with the other histological and immunohistochemical findings, conclusively established the diagnosis of DDLPS with IMT-like features, underscoring the tumor’s aggressive biological behavior and diagnostic complexity. According to the classification system of the French Fédération Nationale des Centres de Lutte Contre le Cancer (FNCLCC), the tumor was classified as grade 2 (G2).
Discussion
This case highlights the diagnostic and clinical challenges posed by DDLPS with IMT-like features, emphasizing the need for a multimodal diagnostic approach. The mesenteric location of this DDLPS adds another layer of rarity, as DDLPS most commonly occurs in the retroperitoneum or extremities [1, 4–9]. The dedifferentiated component in this case exhibited histological features closely mimicking IMT, including myxoid stroma and dense inflammatory infiltrates. This pattern is consistent with the IMT-like variant of DDLPS described in the literature [11, 12]. Other histological patterns of DDLPS have been observed, including myxofibrosarcoma-like, leiomyogenic, osteosarcomatous, chondrosarcomatous, angiosarcomatous, rhabdomyosarcomatous, neural-like or meningothelial-like whorling patterns [1, 5, 21–23]. The diagnostic challenge lies in distinguishing IMT-like DDLPS from other entities. Although the histology is similar, in IMT, frequently ALK, ROS1 or pan-Trk expression can be demonstrated by immunohistochemistry as indicator of an underlying ALK, ROS1 or NTRK gene rearrangement [18]. Desmoid fibromatosis typically exhibits bland fibroblastic spindle cells in a collagen-rich stroma, lacks a dense inflammatory infiltrate and shows immunohistochemical nuclear ß-catenin expression. Tumors cells lack nuclear atypia and MDM2 amplification as seen in DDLPS. The detection of MDM2 cluster amplification as in this case is a hallmark of DDLPS, aiding in its distinction from benign and low-grade mimickers, such as atypical spindle cell/pleomorphic lipomatous tumor or solitary fibrous tumor. Performing MDM2-FISH analysis on a mesenchymal spindle cell tumor in the abdomen or the retroperitoneum is essential for the definitive exclusion of liposarcoma. However, this molecular finding does not assist in distinguishing DDLPS from ALT/WDLPS, as both sarcomas exhibit amplification of MDM2 and most often CDK4. While morphological differentiation is crucial in this context, it is often challenging, particularly in the sclerosing and inflammatory subtypes. Sclerosing ALT/WDLPS is characterized by scattered bizarre stromal cells with hyperchromatic nuclei within a dense collagenous stroma, while inflammatory ALT/WDLPS features chronic inflammatory infiltrates and bizarre multinucleated stromal cells [24–26]. A key feature distinguishing ALT/WDLPS from DDLPS is the presence of mature adipocytes, that typically predominate in ALT/WDLPS but are found only focally in DDLPS [23]. Conversely, DDLPS presents with a dedifferentiated, typically non-lipogenic, low or high-grade morphology that may even overgrow any residual lipogenic features [1, 2]. Furthermore, Harry Evans proposed a mitotic count of ≥ 5 mitoses per 10 HPF as a valid criterion for differentiating DDLPS from ALT/WDLPS, which aligns with our observation of up to 7 mitotic figures per 10 HPF [4, 27].
DDLPS is an aggressive tumor with a high rate of local recurrence and a significant risk of distant metastasis. It is graded as G2 or G3 according to the FNCLCC grading system [28]. Furthermore, myogenic differentiation may be associated with a worse outcome [28]. In the presented case, recurrent tumors developed within two years of initial diagnosis in 2022, consistent with the unfavourable reported clinical behavior of mesenteric DDLPS [11]. Complete surgical resection remains the fundamental treatment approach. Nevertheless, achieving negative margins in mesenteric regions is frequently challenging due to the complex anatomy, substantial tumor burden, and infiltration of surrounding structures and organs [12, 29–31]. The available evidence indicates that radiation therapy may potentially reduce the risk of local recurrence in DDLPS. Nevertheless, the effect of this treatment on overall survival remains unclear [32–34]. The role of adjunctive chemotherapy is still a topic of debate within the scientific community [31, 32, 35, 36].
The prognosis for DDLPS is variable and dependent on a number of factors, including tumor grade, location (with retroperitoneal lesions exhibiting the poorest clinical outcome) and resectability [28]. Five-year survival rates range between 20–40% [29, 30]. The IMT-like variant does not appear to alter the overall prognosis significantly but complicates the diagnostic process, increasing the risk of misdiagnosis and delayed treatment [15, 37].
Conclusion
In conclusion, the IMT-like variant of DDLPS represents a diagnostic pitfall that requires careful histopathological and molecular evaluation. This case underscores the importance of an integrated diagnostic approach and highlights the need for continued research into this aggressive tumor subtype.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Lucas, David R. MD*; Shukla, Abhishek MD*; Thomas, Dafydd G. MD, Ph D*; Patel, Rajiv M. MD*; Kubat, Anthony J. MD†; Mc Hugh, Jonathan B. MD*. Dedifferentiated Liposarcoma With Inflammatory Myofibroblastic Tumor-like Features. The American Journal of Surgical Pathology 34(6):p 844–851, June 2010.10.1097/PAS.0b 013e 3181 db 34d 820431481 · doi ↗ · pubmed ↗