Decoding the genomic landscape of Mammaliicoccus sp. RAM2 isolated from flacherie-infected Bombyx mori L
Rittick Mondal, Dipanjan Das, Sujan Paul, Atanu Manna, Trishanjan Biswas, Biraj Sarkar, Shubhajit Shaw, Arka Pratim Chakraborty, Amit Kumar Mandal

TL;DR
This paper presents the draft genome of a Mammaliicoccus sp. RAM2 strain isolated from a sick silkworm in India, aiding in its species identification.
Contribution
The study provides a draft genome sequence for Mammaliicoccus sp. RAM2, supporting accurate species-level identification.
Findings
The draft genome of Mammaliicoccus sp. RAM2 was sequenced from a flacherie-infected Bombyx mori L.
The genome offers genetic insights for precise species-level identification of Mammaliicoccus.
Abstract
Species of the genus Mammaliicoccus are recognized as opportunistic zoonotic pathogens. Here, we report the draft genome sequence of Mammaliicoccus sp. RAM2, isolated from a flacherie-infected Bombyx mori L. (Nistari race) from Raiganj University, India (25.6071° N, 88.1306° E), which offers valuable genetic insights for precise species-level identification.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
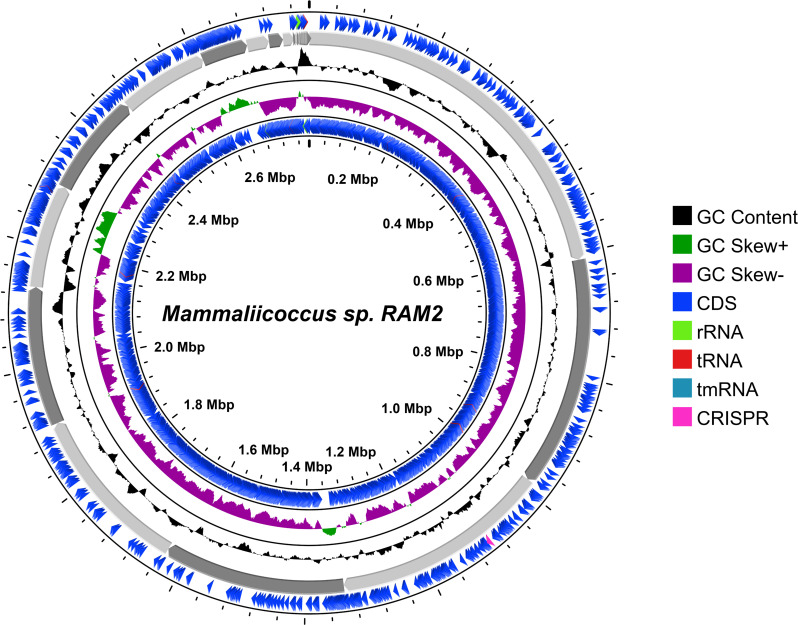 Fig 1
Fig 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsInsect Resistance and Genetics · Vector-borne infectious diseases · Genomics and Phylogenetic Studies
ANNOUNCEMENT
The genus Mammaliicoccus comprises non-motile, gram-positive bacteria that are widely distributed in human samples, agricultural settings, and wildlife habitats. It contributes to antibiotic resistance gene transmission and has been shown to infect silkworms, posing public health risks (1, 2).
Mammaliicoccus sp. strain RAM2 was isolated from the B. mori L larval hemolymph. The collected hemolymph was serially diluted in PBS, spread on mannitol-salt agar (MSA) plate (composition: proteose peptone, 10.0 g; sodium chloride, 75.0 g; D-mannitol, 10.0 g; HM peptone B, 1.0 g, phenol red, 0.025 g, agar, 15.0 g, distilled water, 1 L, pH 7.4) (3) and incubated overnight at 30°C. After incubation, single colonies were picked and transferred to fresh MSA plates to obtain pure cultures. These cultures were maintained on MSA plates for up to 12 generations before being preserved at −80°C in 20% glycerol.
The genomic DNA of RAM2 was extracted by the standard phenol-chloroform method (4). The paired-end libraries were constructed prior to sequencing using the Illumina NovaSeq 6000 platform (Neuberg Diagnostics Pvt. Ltd., Ahmedabad, India). The DNA library was prepared using the KAPA HyperPlus Kit (Roche #07962428001). The final DNA libraries were quantified using the Qubit 4.0 fluorometer (ThermoFisher #Q33238) using the DNA HS assay kit (ThermoFisher #Q32851) following the manufacturer’s protocol. The insert size of the library was identified using the TapeStation 4150 system (Agilent) utilizing highly sensitive D1000 Screentapes (Agilent # 5067–5582) following the manufacturer’s protocol, yielding 23,856,714 reads with a 2 × 150 bp paired-end read length.
Quality assessment of the raw fastq reads of the sample was performed using FastQC v.0.11.9 (default parameters) (5). The raw fastq reads were preprocessed using Fastp v.0.23.4 (6) (parameters: --length_required 50—correction --trim_poly_g --qualified_quality_phred 30—unqualified_percent_limit 30—average_qual 30). Processed data were re-assessed using FastQC. The processed reads were *de novo-*assembled using Unicycler v.0.4.4 (7) with default parameters. Estimations of the completeness and rate of contamination of the assembled genome were done using CheckM2 v.1.0.1 (8). The annotation was carried out via the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) v6.9 with the methods best-placed reference protein set and GeneMarkS-2+ (9). The assembly produced a draft genome sequence encompassing 42 contigs. The N50 value is 294,727 bp, and the L50 count is 4. The estimated genome size is 2,786,715 bp, with a G + C content of 32.39% and 1107.55 x coverage. A total of 2,765 coding sequences were annotated, including three rRNA genes (one 5S, one 16S, and one 23S rRNA genes) and 50 tRNA genes. Finally, a genome map of RAM2 was generated using CGView Server (https://cgview.ca/) (10) (Fig. 1). The presence of antibiotic resistance genes such as salC, sdrM, sepA, vanY (in the vanF cluster), and vanT (in the vanG cluster) within the genome provides insights into the isolate’s genotypic diversity, host–microbe interactions, molecular pathogenesis, and the emerging threat of antimicrobial resistance (AMR). This underscores the need for continued surveillance and the development of novel therapies to combat AMR.
The genome map Mammaliicoccus sp. strain RAM2, generated using the CGView Server. The blue arrows represent CDSs, gray arrows show contigs, the black plot indicates GC content, and the green and magenta plot represents CG skew + and −, respectively.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Crippa BL, de Matos LG, Souza FN, Silva NCC. 2024. Non-aureus Staphylococci and Mammaliicocci (NASM): their role in bovine mastitis and One Health. J Dairy Res 91:44–56. doi:10.1017/S 002202992400016538584301 · doi ↗ · pubmed ↗
- 2Gençer D, Şalvarcı HB, Ulaşlı B, Cengiz FC, Demir İ. 2023. Bacterial diversity associated with the Hatay yellow strain silkworm (Bombyx mori L.): Isolation, identification and characterization. Mustafa Kemal Üniversitesi Tarım Bilimleri Dergisi 28:593–605. doi:10.37908/mkutbd.1288460 · doi ↗
- 3Shields P, Tsang AY. 2006. Mannitol salt agar plates protocols. Available from: https://asm.org/Protocols/Mannitol-Salt-Agar-Plates-Protocols
- 4Marmur J. 1961. A procedure for the isolation of deoxyribonucleic acid from micro-organisms. J Mol Biol 3:208–IN 1. doi:10.1016/S 0022-2836(61)80047-8 · doi ↗
- 5Andrews S. 2010. Fast QC: a quality control tool for high throughput sequence data
- 6Chen S, Zhou Y, Chen Y, Gu J. 2018. Fastp preprocessor. Bioinformatics. doi:10.1093/bioinformatics/bty 560PMC 612928130423086 · doi ↗ · pubmed ↗
- 7Wick RR, Judd LM, Gorrie CL, Holt KE. 2017. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. P Lo S Comput Biol 13:e 1005595. doi:10.1371/journal.pcbi.100559528594827 PMC 5481147 · doi ↗ · pubmed ↗
- 8Chklovski A, Parks DH, Woodcroft BJ, Tyson GW. 2023. Check M 2: a rapid, scalable and accurate tool for assessing microbial genome quality using machine learning. Nat Methods 20:1203–1212. doi:10.1038/s 41592-023-01940-w 37500759 · doi ↗ · pubmed ↗