Complete genome sequence of Escherichia coli phage Eryne
Valentin Druelle, Alexander Harms

TL;DR
This paper presents the full genome sequence of a new Escherichia coli phage called Eryne and classifies it within a specific virus group.
Contribution
The paper provides the complete genome sequence of a newly discovered E. coli phage and classifies it taxonomically.
Findings
The genome of phage Eryne is 145,026 base pairs long.
Eryne belongs to the genus Justusliebigvirus in the Stephanstirmvirinae subfamily.
The phage targets surface glycans of Escherichia coli.
Abstract
Bacteriophage Eryne is a new virus infecting clinical and laboratory strains of Escherichia coli that targets surface glycans. We report the 145,026 bp genome of phage Eryne and show that it belongs to the genus Justusliebigvirus of the Stephanstirmvirinae, known for their multivalent host recognition.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
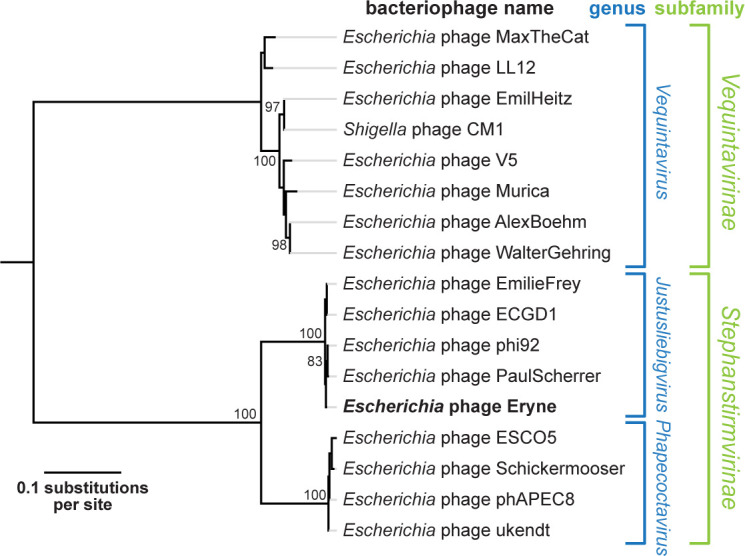 Fig 1
Fig 1- —Schweizerischer Nationalfonds zur Förderung der Wissenschaftlichen Forschunghttp://dx.doi.org/10.13039/501100001711
- —Schweizerischer Nationalfonds zur Förderung der Wissenschaftlichen Forschunghttp://dx.doi.org/10.13039/501100001711
- —Biozentrum, University of Basel
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBacteriophages and microbial interactions · Evolution and Genetic Dynamics · Genomics and Phylogenetic Studies
ANNOUNCEMENT
Phages of genus Justusliebigvirus (phi92-like phages) and their relatives encode several co-expressed tailspikes and tail fibers targeting diverse host receptors, which enable remarkably broad host recognition analogous to a “Swiss Army knife” and hold great potential for biotechnology (1–3). Here, we report the genome of phage Eryne, a new Justusliebigvirus phage. Eryne was isolated in 2020 from wastewater from the local sewage treatment plant of the city of Basel (ARA Basel, 47°34′53.6″N 7°36′00.9″E) by plating on a lawn of Escherichia coli strain UTI89 grown in LB soft agar at 37°C as described previously (2). Plaques obtained with this uropathogenic model strain were tested for growth on the E. coli K-12 MG1655 ΔRM with and without restored expression of O-antigen glycans (wbbL(+) genotype [2]). Phage Eryne grew on both strains, forming clear plaques of 1–2 mm diameter on E. coli K-12 with O-antigen and very small, turbid plaques without O-antigen expression. These results suggest that Eryne encodes a tailspike recognizing the O16-type O-antigen of E. coli K-12 but can only poorly recognize the NGR glycan that seems to be targeted by Justusliebigvirus phages on K-12 strains in its absence (4). The phage was passaged three times to ensure a pure, clonal stock before generating a high-titer lysate using double-agar overlays. Briefly, an LB soft agar with the E. coli K-12 MG1655 ΔRM wbbL(+) host was infected with Eryne for confluent lysis and grown for ca. 16 h at 37°C before being scraped off the plate into ca. 15 mL of SM buffer. This suspension was heavily vortexed and cleared from debris by centrifugation at 8,000 g for 5 minutes to generate the phage stock. Subsequently, the genomic DNA of Eryne was extracted from 1 mL of high-titer stock using the Norgen Biotek Phage DNA Isolation Kit. For sequencing at SeqCenter (Pittsburgh, USA), sample libraries were prepared using the protocol of Baym and colleagues (5) with dual 8 bp indices and sequenced on an Illumina NextSeq 550 producing 2 × 151 bp reads. The data were demultiplexed and adapters were removed using bcl2fastq (version 2.20.0.422; Illumina Inc., San Diego, USA) without additional trimming or quality control at this stage (91% of base pairs had a quality score of Q30). We then used Unicycler version 0.5.0 in Illumina-only mode to assemble 100,000 reads randomly subsampled by Seqtk-1.3 (6) from the total of 2,386,786 reads after automated quality control with FastQC (7) and obtained a circular assembly of 145,026 bp with a GC content of 37.4% and a mean coverage of 165× (8). The genome of Eryne was annotated using Pharokka version 1.7.3 and Phold version 0.2.0 with standard settings after which it was linearized with the 5′ end at the start codon of the large terminase gene (9, 10). Using BLAST (version 2.16.0) of the whole genome of Eryne against the non-redundant nucleotide database of NCBI GenBank (release 262) (11, 12), we determined that this phage belongs to the genus Justusliebigvirus of the Stephanstirmvirinae subfamily by showing >98% identity over >98% of the query sequence for known representatives like PaulScherrer (cutoff: 70% identity across genome length [13]). Phylogenetic analyses confirmed the identity of Eryne as a Justusliebigvirus (Fig. 1).
Major capsid protein gene phylogeny of phage Eryne. The major capsid protein genes of phages belonging to Stephanstirmvirinae genus Justusliebigvirus and relatives were identified as best hits of the Eryne ortholog by BLASTn searches against the non-redundant nucleotide database of NCBI GenBank (release 262) and aligned using MAFFT (version 7.490) implemented in Geneious Prime 2024.0.2 with FFT-NS-1 algorithm, 200PAM/k = 2 scoring matrix, gap open penalty 1.53, and offset value 0.123 (default settings; alignment length 1,036 nt) (14). Subsequently, we calculated a maximum likelihood phylogeny based on this alignment using PhyML (version 3.3.20180621) (15) implemented in Geneious Prime 2024.0.2 with the HKY85 substitution model and 100 bootstraps. The phylogeny was rooted between Vequintavirinae and the other phages. Bootstrap values are shown if >70/100. Phage Eryne is highlighted in bold.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Schwarzer D, Buettner FFR, Browning C, Nazarov S, Rabsch W, Bethe A, Oberbeck A, Bowman VD, Stummeyer K, Mühlenhoff M, Leiman PG, Gerardy-Schahn R. 2012. A multivalent adsorption apparatus explains the broad host range of phage phi 92: A comprehensive genomic and structural analysis. J Virol 86:10384–10398. doi:10.1128/JVI.00801-1222787233 PMC 3457257 · doi ↗ · pubmed ↗
- 2Maffei E, Shaidullina A, Burkolter M, Heyer Y, Estermann F, Druelle V, Sauer P, Willi L, Michaelis S, Hilbi H, Thaler DS, Harms A. 2021. Systematic exploration of Escherichia coli phage-host interactions with the BASEL phage collection. P Lo S Biol 19:e 3001424. doi:10.1371/journal.pbio.300142434784345 PMC 8594841 · doi ↗ · pubmed ↗
- 3Tsonos J, Adriaenssens EM, Klumpp J, Hernalsteens J-P, Lavigne R, De Greve H. 2012. Complete genome sequence of the novel Escherichia coli phage ph APEC 8. J Virol 86:13117–13118. doi:10.1128/JVI.02374-1223118449 PMC 3497662 · doi ↗ · pubmed ↗
- 4Sellner B, Prakapaitė R, van Berkum M, Heinemann M, Harms A, Jenal U. 2021. A new sugar for an old phage: a c-di-GMP-dependent polysaccharide pathway sensitizes Escherichia coli for bacteriophage infection. M Bio 12:e 0324621. doi:10.1128/mbio.03246-2134903045 PMC 8669472 · doi ↗ · pubmed ↗
- 5Baym M, Kryazhimskiy S, Lieberman TD, Chung H, Desai MM, Kishony R. 2015. Inexpensive multiplexed library preparation for megabase-sized genomes. P Lo S One 10:e 0128036. doi:10.1371/journal.pone.012803626000737 PMC 4441430 · doi ↗ · pubmed ↗
- 6Li H. 2018. Seqtk-1.3 - Toolkit for processing sequences in FASTA/Q formats. Available from: https://github.com/lh 3/seqtk
- 7Anderson S. 2023. Fast QC: a quality control tool for high throughput sequence data. Available from: https://qubeshub.org/resources/fastqc
- 8Wick RR, Judd LM, Gorrie CL, Holt KE. 2017. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. P Lo S Comput Biol 13:e 1005595. doi:10.1371/journal.pcbi.100559528594827 PMC 5481147 · doi ↗ · pubmed ↗