Genome sequences of four A1 subcluster Mycobacterium smegmatis bacteriophages
Spencer T. Payne, Jayden S. Longhurst, Elisa A. Correa Lazaro, Matthew N. Jackson, Jacob D. Scott, Sophie B. Daines, Cade B. Brink, Hayzen H. Chamberlain, Jacob D. Gwilliam, George Higgins, Monterey D. Domike, Rachel E. Moffat, Abbey R. Larson, Payson C. Danielson, Hyunbi Hwang

TL;DR
This paper presents the genome sequences of four bacteriophages that infect a specific strain of Mycobacterium smegmatis.
Contribution
The study identifies six novel open reading frames in the genomes of four A1 subcluster bacteriophages.
Findings
Payneful, Marchy, Hami1, and Sorpresa are A1 subcluster phages infecting Mycobacterium smegmatis mc2155.
The phages have genome lengths and guanine–cytosine content consistent with other A1 subcluster phages.
Six novel open reading frames were identified in the genomes of these phages.
Abstract
Payneful, Marchy, Hami1, and Sorpresa are A1 subcluster tailed bacteriophages belonging to the Caudoviricetes class that infect Mycobacterium smegmatis strain mc2155. They are consistent with other A1 subcluster phages based on their genome length and guanine–cytosine content. Their genomes contain six novel open reading frames.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
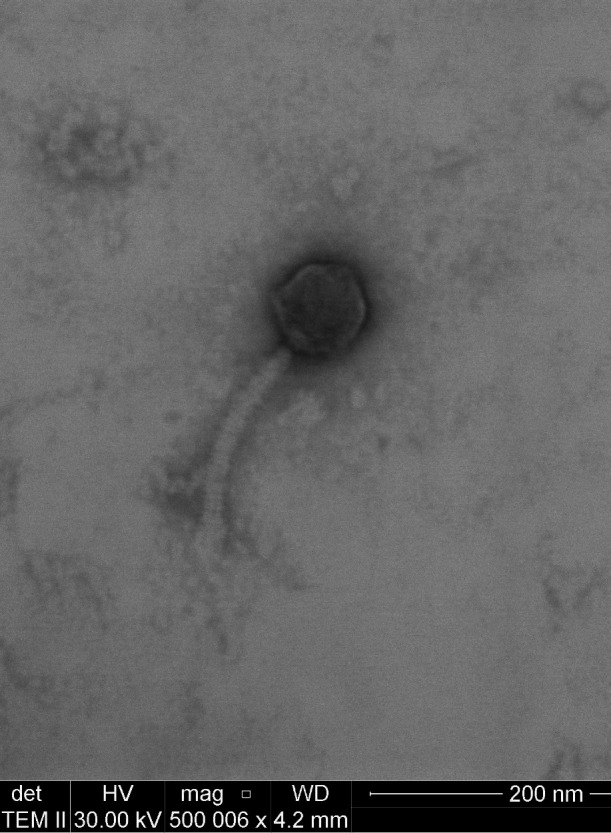 Fig 1
Fig 1| Phage name | Hami1 | Marchy | Payneful | Sorpresa |
|---|---|---|---|---|
| GPS coordinates | 40.47223 N, 111.78163 W | 40.262447 N, 111.65287 W | 40.24018 N, 111.657454 W | 40.27669 N, 111.645 W |
| Isolation details | 2023 | 2023 | 2024 | 2023 |
| Sequence reads | 2.62 | 3.31 | 3.41 | 2.52 |
| Sequence depth | 2,654 | 4,941 | 4,807 | 3,642 |
| Genome length (base pairs) | 47,823 | 50,273 | 49,415 | 52,066 |
| Guanine–cytosine content (%) | 63.5 | 62.7 | 63.8 | 63.2 |
| Number of ORFs | 92 | 87 | 85 | 93 |
| Number of Orphams | 3 | 1 | 1 | 1 |
| Number of ORFs with putative function | 41 | 41 | 39 | 38 |
| Number of ORFs with no putative function | 51 | 46 | 46 | 55 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBacteriophages and microbial interactions · Genomics and Phylogenetic Studies · Mycobacterium research and diagnosis
ANNOUNCEMENT
Bacteriophages continue to be a substantial source of advancements in molecular biology, ranging from host restriction enzymes to CRISPR-Cas systems (1). Additionally, mycobacteriophages have the potential to be used as therapeutics, particularly as an alternative to antimicrobial agents, due to their ability to infect mycobacterial species (2).
The phages reported in this study were isolated from soil samples collected in Provo, Utah. Each sample was suspended in 7H9 broth and filtered through a 0.45 µm filter. An aliquot was used to infect Mycobacterium smegmatis mc^2^155 host cells mixed with top agar, then plated on 7H10 agar prior to incubation at 37°C for 2 days. Resulting plaques (ranging from 2.5 to 4.0 mm with cleared centers and cloudy outer edges) were picked using a sterile micropipette tip. Following at least four rounds of plaque purification, a high titer lysate (>1E10^9^ PFU/mL) was prepared by flooding nearly confluent “web-plates” with Middlebrook 7H9 broth, incubating for 2 h at room temperature, decanting, and filtering through a 0.2 µm filter. DNA was extracted from the lysate by using the Norgen Phage DNA Isolation Kit following the manufacturer’s protocol prior to library preparation and sequencing. Briefly, 0.5 µg of DNA per sample was used for DNA library preparation using the NEBNext Ultra DNA Library Prep Kit for Illumina (NEB, USA) following manufacturer’s recommendations, and unique indices were added to each sample. The DNA samples were sonicated to a size of 350 bp, then DNA fragments were end-polished, A-tailed, and ligated with the full-length adaptor for Illumina sequencing with further PCR amplification. PCR products were purified (AMPure XP System) and libraries were analyzed for size distribution with an Agilent 2100 Bioanalyzer and quantified using real-time PCR. The whole genomes were sequenced using Illumina NovaSeq X PE150 with the corresponding sequencing kit, producing 2.5–3.5 million 150 bp paired-end reads per genome (Table 1). Reads were trimmed using TrimGalore v0.6.6 (3) with a minimum length of 20 bases and a minimum Phred quality score of 20. Trimmed reads underwent assembly with either Unicycler version 0.5.0 (4) or Newbler v2.9 (5) prior to validation using CONSED v2.9 (6) with default parameters. Phages were visualized using negatively stained (2% uranyl acetate) transmission electron micrograph (Fig. 1).
Negative-stained transmission electron micrograph of the Payneful phage (500,000× magnification, 30 kV accelerating voltage, Tecnai TF-20) representative of these four phages.
Assembled fasta files were loaded into DNA Master v5.23.6 (7) where ORFs were called with data generated by Glimmer (8) and GeneMark (9), BLASTN (10), HHpred (11, 12), GeneMarkS (13), Starterator (14), and Phamerator (15). Functional assignments were made by comparison to the same BLAST and HHpred hits, as well as to the Starterator and Phamerator maps (15). Default parameters were used for all tools. The same tools were used to determine the presence of coding regions in large gaps between ORFs for ORFs missed by automated annotation, and in some cases, ORFs were manually added. tRNA analysis was performed using PhageScope (16) and tRNAscan-SE (17). These genomes were assigned to cluster A1 through best-match nucleotide sequence homology searches (95.94% identity, 84% coverage with A1 phage Maroc7).
Six orphams defined as no BLAST hit with e-values < 1e−5 were found across these four phages. The guanine–cytosine content for the phages was consistent with that of their host and other A1 subcluster phages, ranging from 62.7 to 63.8%. No tRNAs were found in these genomes, although ORF 68 of Sorpresa appeared to encode for a putative transposase.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Ofir G, Sorek R. 2018. Contemporary phage biology: from classic models to new insights. Cell 172:1260–1270. doi:10.1016/j.cell.2017.10.04529522746 · doi ↗ · pubmed ↗
- 2Allué-Guardia A, Saranathan R, Chan J, Torrelles JB. 2021. Mycobacteriophages as potential therapeutic agents against drug-resistant tuberculosis. Int J Mol Sci 22:735. doi:10.3390/ijms 2202073533450990 PMC 7828454 · doi ↗ · pubmed ↗
- 3Git Hub. Felix Krueger/Trim Galore: a wrapper around cutadapt and fastqc to consistently apply adapter and quality trimming to fastq files, with extra functionality for RRBS data. Available from: https://github.com/Felix Krueger/Trim Galore
- 4Wick RR, Judd LM, Gorrie CL, Holt KE. 2017. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. P Lo S Comput Biol 13:e 1005595. doi:10.1371/journal.pcbi.100559528594827 PMC 5481147 · doi ↗ · pubmed ↗
- 5Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen Y-J, Chen Z, et al.. 2005. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380. doi:10.1038/nature 0395916056220 PMC 1464427 · doi ↗ · pubmed ↗
- 6Gordon D, Green P. 2013. Consed: a graphical editor for next-generation sequencing. Bioinformatics 29:2936–2937. doi:10.1093/bioinformatics/btt 51523995391 PMC 3810858 · doi ↗ · pubmed ↗
- 7Pope WH, Jacobs-Sera D. 2018. Annotation of bacteriophage genome sequences using DNA Master: an overview. Methods Mol Biol 1681:217–229. doi:10.1007/978-1-4939-7343-9_1629134598 · doi ↗ · pubmed ↗
- 8Delcher AL, Bratke KA, Powers EC, Salzberg SL. 2007. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23:673–679. doi:10.1093/bioinformatics/btm 00917237039 PMC 2387122 · doi ↗ · pubmed ↗