Clinical Exome Sequencing in Pediatric Patients
Orhan Görükmez, Özlem Görükmez, Ali Topak, Hanife Ayşegül Arsoy

TL;DR
This study shows that clinical exome sequencing can effectively diagnose 60% of childhood genetic diseases and identifies 30 new mutations.
Contribution
The study contributes 30 novel mutations to the understanding of genetic disorder profiles in pediatric patients.
Findings
CES detected significant variants in 60% of 68 pediatric patients with suspected genetic disorders.
30 of the 46 identified single nucleotide variants were previously unreported in the literature.
Copy number variations were found in two patients, while the rest had single nucleotide variants.
Abstract
Introduction: The development of genomic sequencing techniques has led to the effective diagnosis of genetic diseases. In this study, clinical exome sequencing (CES) results applied to genetic disorders are reported. Methods: The CES results of pediatric patients with different system involvements and whose complaints were thought to be of genetic origin were evaluated retrospectively. Results: Significant variants associated with complaints were detected in 41 (60%) of 68 patients. Copy number variations were detected in two patients, and single nucleotide variants (SNVs) were detected in the other 39 patients. A total of 46 SNVs were detected in these 39 patients. Sixteen of the detected SNVs were previously reported in the literature, but 30 were novel. Conclusions: This study shows that CES can provide a high diagnosis rate (60%) in childhood genetic diseases. Novel mutations…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
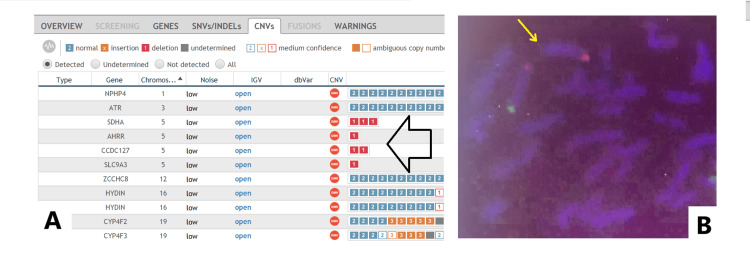 Figure 1
Figure 1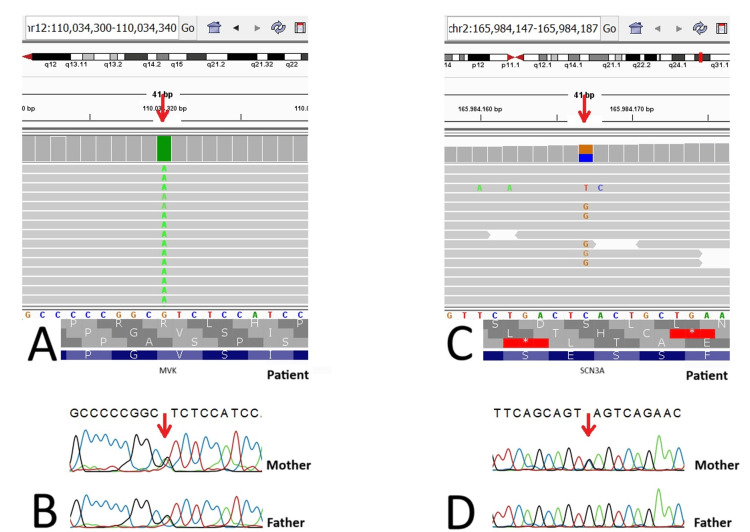 Figure 2
Figure 2| Gene | Variant | Zygosity | Type | R/N | gnomAD | ACMG |
| ARSA | c.827C>T (p.Thr276Met) | Heterozygous | Missense | CM930042 | ||
| c.1393A>G ( | Heterozygous | Missense | N | 0 | UCS | |
| FANCA | c.275C>A ( | Heterozygous | Nonsense | |||
| c.2738A>C (p.His913Pro) | Heterozygous | Missense | ||||
| CHRNA2 | c.1506G>C (p.Arg502Ser) | Heterozygous | Missense | N | 0 | UCS |
| KLF11 | c.1296_1297del ( | Heterozygous | Frameshift | N | 0.0000159 | UCS |
| GALNT12 | c.1281_1296del (p.Trp427Cysfs*23) | Heterozygous | Frameshift | N | 0.00000657 | LP |
| TCOF1 | c.38del (p.Leu13Argfs*18) | Heterozygous | Frameshift | N | 0 | LP |
| P53 NM_000546 | c.733G>A (p.Gly254Ser) | Heterozygous | Missense | CM920674 | ||
| ARFGEF2 | c.5063+2dupT | Homozygous | Splicing | N | 0 | UCS |
| NLRP1 | c.209C>A (p.Ala70Asp) | Heterozygous | Missense | N | 0 | UCS |
| c.3338G>A (p.Arg1113His) | Heterozygous | Missense | N | 0.00000797 | UCS | |
| MVK | c.1129G>A (p.Val377Ile) | Homozygous | Missense | CM990890 | ||
| SCN3A | c.3220G>C (p.Glu1074Gln) | Heterozygous | Missense | N | 0 | UCS |
| ABHD5 | c.594dupC (p.Arg199Glnfs*11) | Homozygous | Frameshift | |||
| TMEM5 | c.1052delC (p.Pro351Leufs*6) | Homozygous | Frameshift | N | 0 | LP |
| RPE65 | c.637C>T (p.Gln213*) | Homozygous | Nonsense | N | 0 | P |
| FAS | c.854A>T (p.His285Leu) | Heterozygous | Missense | N | 0 | UCS |
| TCN2 | c.244C>T (p.Gln82*) | Homozygous | Nonsense | N | 0 | LP |
| AIPL1 | c.683_688delinsA (p.Leu228Hisfs*115) | Homozygous | Frameshift | N | 0 | LP |
| MOCOS | c.2368G>T (p.Glu790*) | Homozygous | Nonsense | N | 0 | LP |
| GBE1 | c.1009T>G (p.Phe337Val) | Homozygous | Missense | N | 0 | UCS |
| ATP6V0A4 | c.2419C>T (p.Arg807*) | Homozygous | Nonsense | N | 0.0000239 | UCS |
| COL1A1 | c.2533G>A (p.Gly845Arg) | Heterozygous | Missense | |||
| GRIK1 | c.332C>A ( | Homozygous | Missense | N | 0 | UCS |
| SH2D1A | c.213_224del (p.Val72_Arg75del) | Hemizygous | Inframe | N | 0 | LP |
| EIF2B4 | c.1409A>C (p.His470Pro) | Homozygous | Missense | N | 0 | UCS |
| ACAD9 | c.797G>A (p.Arg266Gln) | Homozygous | Missense | |||
| HK1 | c.1586T>C (p.Leu529Ser) | Homozygous | Missense | CM950628 | ||
| TRPM6 | c.151C>T (p.Arg51*) | Homozygous | Nonsense | |||
| WNK4 | c.1591_1593del ( | Heterozygous | Inframe | N | 0 | UCS |
| CACNA1A | c.6661_6662ins31 (p.His2221Profs*?) | Heterozygous | Frameshift | N | 0 | LP |
| SCN2A | c.4606A>G ( | Heterozygous | Missense | N | 0 | UCS |
| CDKL5 | c.730T>A (p.Phe244Ile) | Heterozygous | Missense | N | 0 | UCS |
| SLC5A2 | c.655G>A (p.Ala219Thr) | Homozygous | Missense | |||
| ACTA1 | c.282C>A (p.Asn94Lys) | Heterozygous | Missense | CM115391 | ||
| FANCL | c.974C>G (p.Pro325Arg) | Homozygous | Missense | N | 0 | UCS |
| SLC2A9 | c.162+1del | Homozygous | Splicing | N | 0 | P |
| XDH | c.3710C>G (p.Pro1237Arg) | Homozygous | Missense | N | 0.00000398 | UCS |
| IKBKG | c.299C>T (p.Pro100Leu) | Hemizygous | Missense | N | 0 | UCS |
| INPP5E | c.1697G>T (p.Gly566Val) | Homozygous | Missense | N | 0 | UCS |
| BBS7 | c.712_715delAGAG (p.Arg238Glufs*59) | Homozygous | Frameshift | CD093973 | ||
| ASS1 | c.970+5G>A | Heterozygous | Splicing | |||
| c.1168G>A (p.Gly390Arg) | Heterozygous | Missense | CM900037 | |||
| CEP290 | c.5557C>T (p.Gln1853*) | Heterozygous | Nonsense | N | 0 | LP |
| c.5668G>T (p.Gly1890*) | Heterozygous | Nonsense | ||||
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Rare Diseases · Genetics and Neurodevelopmental Disorders · Genomic variations and chromosomal abnormalities
Introduction
It is assumed that around 300 million people worldwide have an orphan disease, the vast majority of which is of genetic origin [1]. With the increasing applicability of genomic sequencing technology, the molecular basis of more than 7100 genetic diseases has been identified in the Online Mendelian Inheritance in Man (OMIM) database [2]. Gene panels, clinical exome sequencing, and whole exome and genome sequencing are among the next-generation sequencing (NGS) techniques that are routinely applied today [3]. Approximately 85% of the variants that cause diseases are found in exons and splicing regions, and exome tests that enable sequencing of these regions have gained importance. Clinical exome sequencing (CES), designed to sequence exons and splice regions of genes with known phenotypic effects, is frequently used as a routine diagnostic test [4]. In this study, we aimed to present the results of the CES test in pediatric patients from 67 different families together with their clinical findings.
Materials and methods
This retrospective study examined clinical findings and molecular genetic results of pediatric patients who presented to the Department of Medical Genetics at Bursa Yüksek İhtisas Training and Research Hospital, Bursa, between 2019 and 2022. The data were meticulously retrieved and curated from archived clinical and molecular records. Ethical approval for the study was obtained from the Ethics Committee of Bursa Yüksek İhtisas Training and Research Hospital (Approval No: 2011-KAEK-25 2022/11-07). The findings presented in this study were systematically derived from molecular analyses and bioinformatics evaluations, as described below in detail.
DNA samples were isolated from the peripheral blood of patients using a QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). A total of 4493 genes were amplified from patient DNA samples using a CES kit (Clinical Exome Solution v2, Sophia Genetics SA., Saint Sulpice, Switzerland) on the NGS platform (NextSeq 500 system, Illumina, San Diego, California, USA), per the manufacturers’ instructions. The Sophia Data Driven Medicine (DDM^TM)^ platform (Sophia Genetics SA., Saint Sulpice, Switzerland) was used for bioinformatics analysis. All analyses of single nucleotide variants (SNVs), insertions and deletions (indels), and copy number variations (CNVs) were performed using this platform. In particular, variants with a minor allele frequency (MAF) <1%, associated with the clinical findings of the patients, were considered. Capillary electrophoresis was used to confirm variants that were considered significant and for segregation analysis. Primers for the variant-associated target sites were designed using Primer 3 (https://primer3.org/). A polymerase chain reaction (PCR) was performed using custom primers and H Taq polymerase (Zeydanlı, Ankara, Turkey) on a thermal cycler (Applied Biosystems^TM^ Veriti^TM^, Thermo Fisher Scientific, USA). Purification of the amplified products was carried out using a Zymo Research Sequencing Clean-up Kit (Epigenetic Companies, Irvine, CA, USA). Cycle sequencing was conducted using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems®, Thermo Fisher Scientific, USA) on a 3500 Genetic Analyzer (Applied Biosystems®, Thermo Fisher Scientific, USA). The sequenced regions were analyzed using CLC Main Workbench v8.1 software (Qiagen).
Confirmation was obtained by fluorescence in situ hybridization (FISH) for the patient with heterozygous deletion of consecutive genes in the 5p subtelomere region in CNV analysis. A peripheral blood sample was cultured in RPMI-1640 medium supplemented with fetal bovine serum (FBS) and phytohemagglutinin (PHA). To obtain metaphase chromosomes, colcemid (0.1 µg/mL) was applied, followed by hypotonic treatment with 0.075 M KCl (37°C, 20 min). The cells were then fixed with a methanol:acetic acid solution (3:1). Genetic alterations associated with cri du chat syndrome were investigated using a specific fluorescent probe (Vysis ToTelVysion Multi-Color FISH Probe, Abbott Molecular, Des Plaines, IL, USA). Preparations were denatured at 75°C for two minutes and hybridized overnight at 37°C on a Leica ThermoBrite device (Leica Biosystems, Germany). Post-hybridization washes were performed using 0.4× saline-sodium citrate buffer (SSC)/0.3% nonidet P-40 (NP-40) (72°C), followed by a final wash with 2× SSC/0.1% NP-40 at room temperature. DAPI or 4',6-diamidino-2-phenylindole staining was applied to visualize nuclear structures. Fluorescent signals were analyzed using a fluorescence microscope (Allegro Plus, BioView, Rehovot, Israel), and at least 20 metaphases were assessed for the presence of 5p deletion.
Information was obtained on whether the variants were reported in the literature using the Human Gene Mutation Database (HGMD) [5]. Variants not reported in the literature at the time of this study were considered novel. The frequency of novel variants in the population was determined (gnomAD) [6] and categorized according to the American College of Medical Genetics and Genomics (ACMG) criteria [7]. Disease-related syndromes were determined in the OMIM database by evaluating the symptoms, family histories, examination findings, and detected variants.
Results
This study was conducted on pediatric patients whose anamnesis, clinical complaints, and physical examination findings indicated a potential genetic basis for their condition. All data utilized in the study were meticulously compiled retrospectively from archived clinical and molecular records to ensure a comprehensive and systematic evaluation. Of the 68 patients included in the study, 39 were male, and 29 were female. Variants associated with clinical findings were detected in 41 patients: CNV in two patients and 46 SNVs in 39 patients (Table 1). One patient with SNV also had a mitochondrial DNA (mtDNA) mutation. In addition, an incidental variant was detected in another patient with an SNV. No variants that could cause disease were detected in the remaining 27 patients. A total of 46 different SNVs were detected, including the incidental one. Of the SNVs, 16 were previously reported in the literature, and 30 were novel. Novel variants were classified according to ACMG criteria; it was seen that 19 of them were of uncertain significance (UCS), nine of them were likely pathogenic (LP), and two of them were pathogenic (P). In addition, 25 of the SNVs were missense, eight were nonsense, eight were frameshift, three were splicing, and two were in-frame types. One of the CNVs was a partial homozygous deletion in intron 12 of the *DPYD *gene. The other mutation was a heterozygous deletion of more than one gene localized in the 5p region. Of all the variations, seven were de novo, and 40 were inherited from at least one parent. Segregation analysis could not be performed for these two variants.
According to the Human Phenotype Ontology (HPO) filtering, connective tissue (n=1), craniofacial (n=1), gastrointestinal (n=2), genitourinary (n=1), hematological (n=13), immune (n=5), metabolic (n=6), mitochondrial (n=1), neurological (n=10), neurometabolic (n=3), neuromuscular (n=3), ophthalmic (n=1), and renal (n=7) diseases were detected. In addition, seven patients had multiple system disorders, five had a predisposition to cancer, and two had a complex phenotype. The incidental variant that we detected in one of our patients as having the potential to affect the endocrine system was not included in the HPO. In one of our patients, the incidental variant, which we thought was the cause of the maturity-onset diabetes of the young type 7 (MODY7), was not included in the HPO.
In the study group, there was a consanguineous marriage between the parents of 25 of our patients, and only two of these patients could not be diagnosed. Regardless of the consanguineous marriage, 14 of our patients had family members with the same molecular diagnosis or similar symptoms.
In this study, 44 different syndromes reported in the OMIM database were identified in our patients. A phenotype associated with the *GRIK1 *variant detected in one of our patients has not yet been reported in the OMIM database. We know that 5p deletion syndrome is not inherited, and most other syndromes show an autosomal-recessive inheritance pattern (26/44). Others were autosomal-dominant (11/44) and X-linked recessive (2/44) inheritance types. An inheritance pattern for *GRIK1 *defect, MODY7, colorectal cancer susceptibility to one was not specified in OMIM. Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) show a mitochondrial inheritance pattern.
Discussion
This investigation centered on pediatric patients presenting with suspected genetic disorders who underwent NGS, integrating their clinical findings, familial histories, and physical examination data. The study synthesizes the demographic characteristics, clinical presentations, and molecular diagnostic outcomes of these patients. Specific cases are detailed to highlight unique findings and illustrate the diverse genetic etiologies observed in the cohort.
All variants detected in our patients were associated with different genes. Therefore, we did not have any two patients with the same diagnosis. There were individuals in a family with different diagnoses. Cases 53 and 54, with complaints of seizures, were siblings (Appendix 1). The male patient was born from a naturally conceived pregnancy, and the female patient was born from an in vitro fertilization (IVF) pregnancy. The variant (SCN2A, c.4606A>G, p․Ser1536Gly) detected in the male patient was inherited from his father, who had similar childhood complaints. The variant (CDKL5, c.730T>A, p.Phe244Ile) detected in the sister from the IVF pregnancy was de novo in a different gene. This situation raised the question, "Does the frequency of de novo mutations increase due to the IVF procedure?" There is no statistically significant difference in the incidence of de novo mutations between individuals born from medical-assisted reproductive pregnancies and those born from naturally conceived pregnancies [8]. A homozygous aryl hydrocarbon receptor interacting protein-like 1 (AIPL1) pathogenic variant was detected in case 35, who had congenital blindness. Since the patient also had findings suggestive of a neurometabolic disease, a whole mtDNA analysis was performed. We also detected the pathogenic variant m.5543T>C, which is compatible with MELAS. The coexistence of nuclear and mtDNA mutations has been reported rarely and mostly at the case level in the literature. We did not find the coexistence of these two diseases detected in our case [9-11].
In our study, we diagnosed two patients using CNV analysis. Performing CNV analyses using exome sequencing has increased the diagnosis rate. It can provide an idea for small regions that cannot be detected using the microarray method [12]. We detected that the CCDC127, SDHA, AHRR, and *SLC9A3 *genes localized in the 5p15.33 region, respectively, were in a single copy number in an eight-year-old male patient numbered 29. We could not clarify the boundaries of the deletion because we could not apply MicroArray to the patient owing to technical limitations. We confirmed the deletion using a 5p subtelomeric FISH probe (Figure 1). The 5p deletion syndrome, also known as cri du chat syndrome, has a very heterogeneous phenotype due to differences in breakpoints [13]. Dysmorphic findings were indistinct, but the neurological symptoms were prominent. In the cranial magnetic resonance imaging of the patient, who was followed up for intellectual development and speech disorder, enlargement of the posterior horns of the lateral ventricles and periventricular hyperintensity were observed. The patient, who was followed up with a prediagnosis of hypoxic-ischemic encephalopathy until the age of eight years, was diagnosed with 5p deletion syndrome following CES. Another patient with CNV change was female (case 26). A section of intron 12 in the *DPYD *gene could not be sequenced. The fact that the CES kit covers this intronic region is advantageous. We detected a homozygous deletion of 380 bp by Sanger sequencing using custom primers. Dihydropyrimidine dehydrogenase deficiency caused by *DPYD *gene mutations can cause multiple organ disorders, mainly in the neurological system [14]. Our 15-month-old female patient had severe neurological problems and vision loss.
Detection of 5p15.33 deletion in the cri du chat syndrome patient by CNV analysis and FISHA: Copy number variation (CNV) analysis shows a heterozygous deletion in the 5p15.33 region, affecting the SDHA, AHRR, CCDC127, and SLC9A3 genes, which are indicated with a black arrow. B: Fluorescence in situ hybridization (FISH) image of a metaphase cell. The 5q telomeric region is marked with an orange signal, while the 5p telomeric region is labeled with a green signal. The deleted 5p telomeric region is indicated by a yellow arrow.
A homozygous missense variant (c.332C>A/p. Ser111Tyr) in the *GRIK1 *gene was detected in a four-month-old female infant (case 43) with severe neurometabolic and retinal problems. *GRIK1 *is among the genes that have not yet been definitively associated with a specific phenotype in the OMIM database. Chatterjee et al. associated *GRIK1 *gene alterations with attention deficit hyperactivity disorder [15], and Forero DA stated that it was a candidate gene for epilepsy [16]. This variant could not fully explain our patient's problems other than epilepsy; therefore, we suggested to her family that more comprehensive tests, such as whole genome sequencing (WGS), should be performed on the patient. Whether this change is an infrequent polymorphism or a disease factor will become clear as the number of cases increases.
In cases where secondary findings were detected in clinical exome studies, the method to be followed was determined according to ACMG criteria. It is recommended that the LP and P secondary variants of the *HNF1A *and *HNF1B *genes cause types 3 and 5, respectively, in the MODY group [17]. We detected a frameshift variant in *KLF11 *in addition to the *CHRNA2 *variant, which is the cause of the disease, in a six-year-old female patient (case 4) who underwent CES for febrile seizure complaints. *KLF11 *has been associated with MODY type 7 in OMIM, but its inheritance pattern is not specified (https://omim.org/). Notably, Laver et al. argued that the *KLF11 *gene is not associated with MODY in a study they conducted [18]. It is evident that this situation will become clear with the acceleration of functional studies and an increase in the number of cases. The *KLF11 *variant detected in our patient was inherited from the mother. Although the clinical significance of this variant is unknown according to the ACMG criteria, it has been reported that the mother was also diagnosed with diabetes mellitus at a young age. The patient was referred to the Pediatric Endocrinology Department.
The detection of the homozygous pathogenic *RPE65 *variant associated with Leber congenital amaurosis 2 (MIM:204100) in an 11-month-old male patient (case 30) is important in terms of treatment. Molecular diagnosis of diseases of genetic origin is undoubtedly of great importance in guiding disease treatment. Since 1990, when the first gene therapy was applied, studies have been conducted on gene therapy in many genetic diseases [19]. One of these diseases is Leber congenital amaurosis, which is caused by *RPE65 *mutations and has been approved for treatment [20]. Therefore, the patient was referred to the ophthalmology unit.
More than one molecular pathology affecting the phenotype can be detected in comprehensive genetic tests such as CES [21]. Case 25 with the *MVK *pathogenic variant associated with hyper-IgD syndrome (MIM:260920) also carried the maternally inherited *SCN3A *variant (Figure 2). Although its clinical significance was uncertain, this variant was important to report since the mother had a history of childhood epilepsy.
Genetic analysis of MVK and SCN3A variants in the patient and parentsA: Integrated genomics viewer (IGV) visualization of the homozygous MVK; c.1129G>A; (p.Val377Ile) variant in the patient; B: Sanger sequencing chromatograms of the heterozygous MVK; c.1129G>A; (p.Val377Ile) variant in the parents; C: IGV track of the heterozygous SCN3A; c.3220G>C; (p.Glu1074Gln) variant in the patient, shown in the forward strand; D: Sanger sequencing results displaying the heterozygous SCN3A; c.3220G>C; (p.Glu1074Gln) variant in the mother and the wild-type sequence in the father, visualized in the reverse strand. The variant positions are indicated by red arrows.
In our study, which included the pediatric age group, the diagnosis rate was 60%. We attribute our relatively high diagnosis rate to our preference for CES over a panel of responsible genes in some cases with a specific phenotype but heterogeneous genetic etiology. Our CNV analysis may be another reason. However, when UCS variants are excluded, our diagnosis rate is 35% (24/68). CES has recently been used frequently in the diagnosis of genetic diseases because of its advantages, such as low cost, fast application, and easier analysis [22]. For these reasons, we preferred to apply CES to patients in our laboratory, which operates for diagnostic purposes. While the diagnostic range of CES is 50%-80% for diseases in the neonatal period, this rate decreases to 25%-50% for diseases presenting in adults or older people [22].
A notable limitation of this study is the inability to further investigate variants classified as uncertain significance (UCS) according to ACMG criteria. This limitation prevented the clarification of these variants, which might have provided additional insights into the molecular diagnoses. The other limitation of this study is that we were unable to perform MicroArray analysis to determine the exact breakpoints of the CNV detected in case 29 due to technical constraints.
We included 27 patients (~40%) who could not be diagnosed, and more extensive tests, such as whole-exome sequencing (WES) or WGS, were offered to these patients.
Conclusions
Thirty novel variants associated with different syndromes were identified in the present study. Emphasis was placed on the clinical features and mutation profiles of genetic diseases, and complex phenotypes with different molecular etiologies must be considered. We believe that our study contributes to the literature in this respect.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Genome sequencing data analysis for rare disease gene discovery Brief Bioinform Umlai UI Bangarusamy DK Estivill X Jithesh PV 363232022 https://doi.org/10.1093/bib/bbab 36310.1093/bib/bbab 36334498682 · doi ↗ · pubmed ↗
- 22022: a pivotal year for diagnosis and treatment of rare genetic diseases Cold Spring Harb Mol Case Stud Kingsmore SF 620482022 https://doi.org/10.1101/mcs.a 00620410.1101/mcs.a 006204 PMC 895890735217563 · doi ↗ · pubmed ↗
- 3Editorial: NGS technologies of rare diseases diagnosis Front Pediatr Couce ML González-Vioque E 1032359102022 https://doi.org/10.3389/fped.2022.10323593644416910.3389/fped.2022.1032359 PMC 9699829 · doi ↗ · pubmed ↗
- 4Five years' experience of the clinical exome sequencing in a Spanish single center Sci Rep Arteche-López A Ávila-Fernández A Riveiro Álvarez R 19209122022 https://doi.org/10.1038/s 41598-022-23786-63635750710.1038/s 41598-022-23786-6PMC 9649665 · doi ↗ · pubmed ↗
- 5HGMD® Professional 2022.4, QIAGEN 3 2024 2022 https://www.hgmd.cf.ac.uk/
- 6Genome Aggregation Database 1 2025 2024 https://gnomad.broadinstitute.org/
- 7Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology Genet Med Richards S Aziz N Bale S 405424172015 https://doi.org/10.1038/gim.2015.302574186810.1038/gim.2015.30PMC 4544753 · doi ↗ · pubmed ↗
- 8De novo mutations in children born after medical assisted reproduction Hum Reprod Smits RM Xavier MJ Oud MS 13601369372022 https://doi.org/10.1093/humrep/deac 0683541311710.1093/humrep/deac 068PMC 9156847 · doi ↗ · pubmed ↗