Haplotype Phasing of Biallelic WNT10B Variants Using Long‐Read Sequencing in Split‐Hand/Foot Malformation Syndrome
Jelena Pozojevic, Naseebullah Kakar, Henrike L. Sczakiel, Nathalie Kruse, Kristian Händler, Saranya Balachandran, Varun Sreenivasan, Martin A. Mensah, Malte Spielmann

TL;DR
Long-read sequencing was used to determine the genetic cause of a rare limb malformation syndrome in a patient with two WNT10B gene variants.
Contribution
Demonstrates the effectiveness of long-read sequencing for haplotype phasing in diagnosing autosomal recessive disorders without parental DNA.
Findings
Long-read sequencing identified two WNT10B variants on separate alleles in a patient with SHFM.
One variant was previously reported, while the other was novel and not found in gnomAD.
Haplotype phasing confirmed the biallelic nature of the variants without parental DNA samples.
Abstract
Split‐hand/foot malformation syndrome (SHFM) is a congenital limb malformation that is both clinically and genetically heterogeneous. Variants in WNT10B are known to cause an autosomal recessive form of SHFM. Here, we report a patient born to unrelated parents who was found to be a compound heterozygote for missense variants in WNT10B: c.994C>T, p.(Arg332Trp) and c.638T>G, p.(Phe213Cys). The variants were identified using long‐read PacBio sequencing, which enabled phasing and confirmed that they were located on different alleles. The maternally inherited variant p.(Arg332Trp) has been previously reported, whereas the paternally inherited variant p.(Phe213Cys) is novel and absent from the gnomAD database. Our findings highlight the utility of long‐read haplotype phasing, which provides valuable insights in determining the biallelic nature of variants in recessive disorders when parental…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
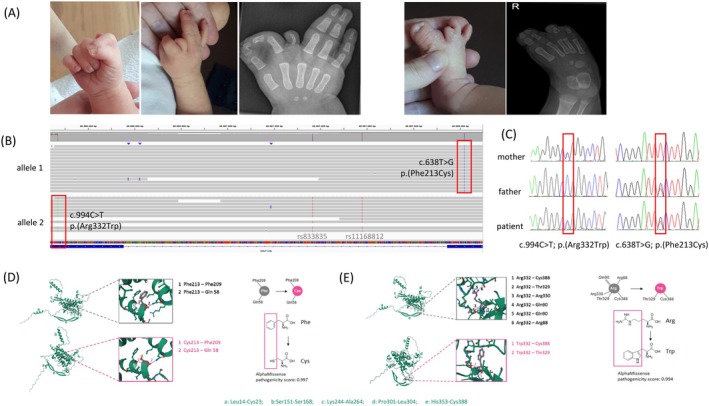 FIGURE 1
FIGURE 1- —Else Kröner‐Fresenius‐Stiftung 10.13039/501100003042
- —Deutsche Forschungsgemeinschaft 10.13039/501100001659
- —Alexander von Humboldt‐Stiftung 10.13039/100005156
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCongenital limb and hand anomalies · Neurogenetic and Muscular Disorders Research · Congenital heart defects research
1
Whole exome sequencing (WES) is the standard technology in clinical genetics for detecting protein‐coding variants. Conversely, whole genome sequencing (WGS) can detect multiple genetic alterations, genome wide. Short‐read technologies, while highly accurate, cannot resolve repetitive and GC‐rich regions, but this problem can be overcome with long‐read sequencing that can resolve difficult sequences and provide epigenetic information.
Split‐hand/foot malformation (SHFM) is a congenital limb condition characterized by genetic heterogeneity, variable expression, and incomplete penetrance. Loci linked to SHFM include TP63, DLX5, DLX6, FGFR1, MAP3K20, UBA2, BHLHA9, and WNT10B [1]. Autosomal recessive, mainly homozygous WNT10B variants cause SHFM6 (OMIM #225300), while heterozygous variants cause dental anomalies [2, 3]. Here, we report a patient with SHFM caused by compound‐heterozygous WNT10B variants, detected using long‐read sequencing. This technology enabled us to phase the reads and prove the variants' biallelic nature.
The patient was born at 37 + 5 weeks with good overall health. The fontanelles, lymph nodes, and genitalia were unremarkable. However, skeletal limb abnormalities were noted. The right hand showed syndactyly of digits I + II and III + IV, with an extra finger between II + III. The left hand had five fingers with a skin bridge between III + IV. Both feet exhibited split malformations: the left foot lacked digit II, had a rudimentary digit III, and syndactyly of IV + V, while the right foot showed syndactyly of I + II and IV + V, with a rudimentary digit III (Figure 1A). Array CGH identified a maternally inherited intronic TP63 deletion (chr3:189647416‐189650138; GRCh38), common in controls and therefore excluded as disease‐causing. Long‐read PacBio HiFi sequencing (average read length: 21081 bp) revealed two WNT10B variants: c.994C>T, p.(Arg332Trp) and c.638T>G, p.(Phe213Cys). Phasing (based on genome‐wide SNVs and indels) confirmed the variants were located on different alleles (Figure 1B). Sanger sequencing validated these findings, showing maternal inheritance of c.994C>T and paternal inheritance of c.638T>G (Figure 1C). The variant c.994C>T was previously reported as homozygous in a consanguineous family with SHFM [2], classified as pathogenic in ClinVar, with a CADD score of 32. According to ACMG criteria [4], it is pathogenic. The c.638T>G variant, absent in gnomAD, has a CADD score of 31 and is classified as likely pathogenic. AlphaFold [5] predictions showed that both variants altered WNT10B protein structure. p.(Phe213Cys) substituted a benzyl group with a thiol side chain, while p.(Arg332Trp) disrupted hydrogen bonds and replaced guanidino group with indole. Both variants were classified as pathogenic by the AlphaMissense pathogenicity score (Figure 1D,E).
(A) Patient's phenotype. (B) Long‐read sequencing showing the biallelic variants and informative SNPs. (C) Sanger sequencing confirming the variants and their parent‐of‐origin. (D, E) In silico predictions of protein folding and hydrogen bonds for wild‐type versus mutant proteins, highlighting structural changes caused by each variant. Variants with AlphaMissense pathogenicity scores > 0.56 are classified as pathogenic.
Biallelic WNT10B variants cause autosomal recessive SHFM, and most cases are homozygous due to consanguinity, but few compound‐heterozygous variants have also been reported. Our patient was born to non‐consanguineous unaffected parents. After confirming that each of these variants was inherited from one parent, we sought to re‐evaluate the clinical status of the parents, since incomplete penetrance and dental anomalies were reported in heterozygous carriers, but they were unavailable.
Short read‐WES could have identified these variants at lower costs, but our work demonstrates the power of long‐reads in proving biallelic inheritance. This can be particularly informative when parental DNA is unavailable, when variants are de novo, or display mosaicism. Haplotype determination cannot be accurately done by short‐read sequencing and traditionally requires extensive experiments. In comparison, it is straightforward with long‐read sequencing to determine whether variants are in trans, supporting a recessive disease phenotype. Although this technology is typically used to resolve difficult sequences (structural variants, GC‐rich and repetitive regions, DNA methylation), we highlight its use in haplotype phasing, which can be critical in arriving at a molecular diagnosis. This study also expands the WNT10B mutational spectrum since c.638T>G has not been previously reported. As sequencing costs decrease, long‐read sequencing might become the preferred technology in clinical genetics, serving as a comprehensive diagnostic test, particularly in the absence of parental samples.
Consent
Written informed consent was obtained.
Conflicts of Interest
The authors declare no conflicts of interest.
Peer Review
The peer review history for this article is available at https://www.webofscience.com/api/gateway/wos/peer‐review/10.1111/cge.14706.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1M. Umairs and A. Hayat , “Nonsyndromic Split‐Hand/Foot Malformation: Recent Classification,” Molecular Syndromology 10, no. 5 (2020): 243–254.32021595 10.1159/000502784 PMC 6997797 · doi ↗ · pubmed ↗
- 2S. A. Ugur and A. Tolun , “Homozygous WNT 10b Mutation and Complex Inheritance in Split‐Hand/Foot Malformation,” Human Molecular Genetics 17, no. 17 (2008): 2644–2653.18515319 10.1093/hmg/ddn 164 · doi ↗ · pubmed ↗
- 3P. Yu , W. Yang , D. Han , et al., “Mutations in WNT 10B Are Identified in Individuals With Oligodontia,” American Journal of Human Genetics 99, no. 1 (2016): 195–201.27321946 10.1016/j.ajhg.2016.05.012PMC 5005437 · doi ↗ · pubmed ↗
- 4S. Richards , N. Aziz , S. Bale , et al., “Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology,” Genetics in Medicine 17, no. 5 (2015): 405–424.25741868 10.1038/gim.2015.30PMC 4544753 · doi ↗ · pubmed ↗
- 5J. Jumper , R. Evans , A. Pritzel , et al., “Highly Accurate Protein Structure Prediction With Alpha Fold,” Nature 596, no. 7873 (2021): 583–589.34265844 10.1038/s 41586-021-03819-2PMC 8371605 · doi ↗ · pubmed ↗