Incontinentia Pigmenti: A Rare Case of Survival of a Male Infant
Riley Shin, Helen Chen, Michelle Tarbox

TL;DR
A rare case of a male infant surviving with incontinentia pigmenti, a typically lethal X-linked disorder, is reported, highlighting unusual genetic factors and the need for ongoing monitoring.
Contribution
The case presents a rare male survival with incontinentia pigmenti and no IKBKG mutations, suggesting somatic mosaicism as a possible mechanism.
Findings
The male infant showed typical skin symptoms of incontinentia pigmenti but no neurological, dental, or ocular issues.
Genetic testing found no IKBKG mutations, indicating a possible somatic mosaicism mechanism for survival.
The case emphasizes the need for multidisciplinary follow-up in male IP patients for potential extracutaneous complications.
Abstract
This case describes a seven-month-old male infant with incontinentia pigmenti (IP), a rare X-linked dominant disorder that is typically lethal in male fetuses. The infant presented with blaschkoid hyperpigmentation but showed no neurologic, dental, or ocular abnormalities. Genetic testing revealed a normal karyotype without IKBKG (inhibitor of nuclear factor kappa B kinase regulatory subunit gamma) mutations, suggesting possible somatic mosaicism, a rare survival mechanism in male patients with IP. This case underscores the rarity of IP in male infants and highlights the need for multidisciplinary follow-up to monitor potential extracutaneous involvement. These findings contribute to the limited understanding of male presentations of IP and their clinical management.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
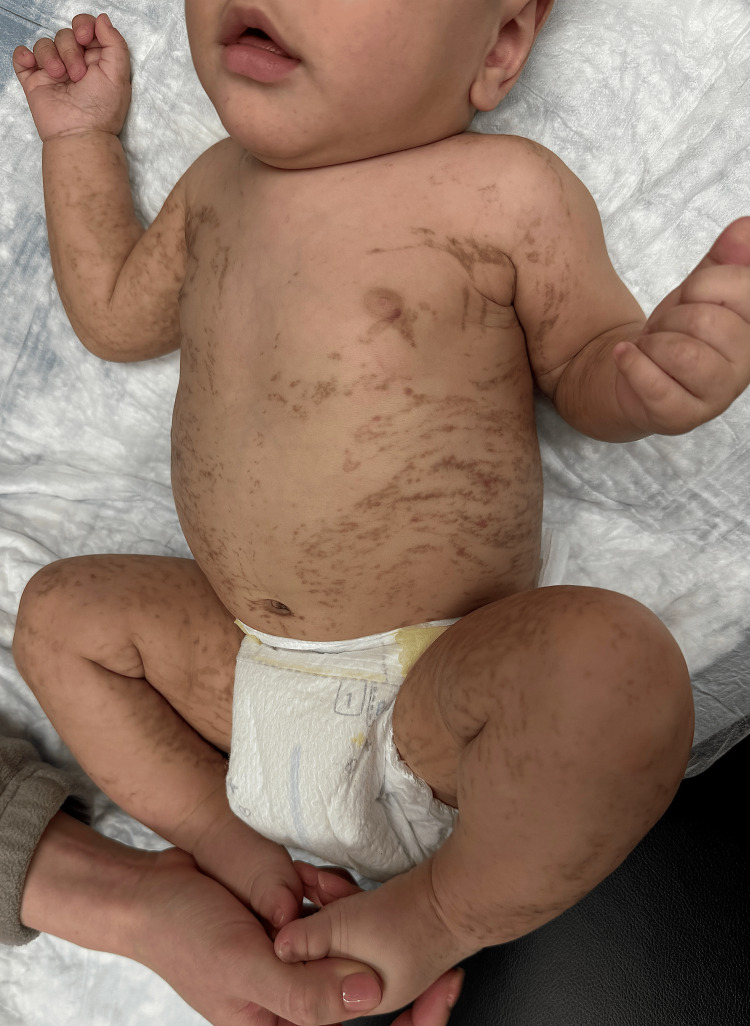 Figure 1
Figure 1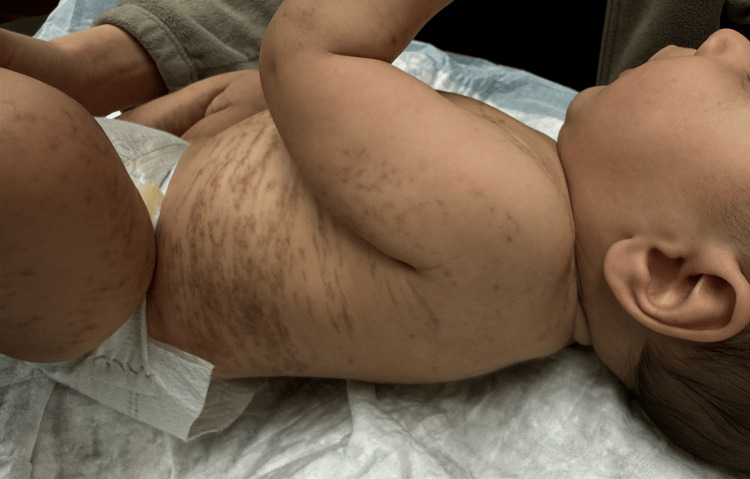 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic and rare skin diseases. · Cancer and Skin Lesions · Hedgehog Signaling Pathway Studies
Introduction
Incontinentia pigmenti (IP), also known as Bloch-Sulzberger syndrome, is a rare genetic disorder caused by a mutation in the IKBKG (inhibitor of nuclear factor kappa B kinase regulatory subunit gamma) gene. This gene encodes NEMO (NF-kappa-B essential modulator), a regulatory protein in the NF-κB (nuclear factor kappa B) signaling pathway necessary for proper embryogenesis. Approximately 80% of IP cases result from deletions of exons 4-10 in the IKBKG gene located on chromosome Xq28. This X-linked dominant mutation occurs in approximately 1 in 50,000 newborns and is typically lethal in male fetuses, with a female predominance of 37:1 [1]. IP is characterized by distinctive cutaneous manifestations, along with abnormalities in the teeth, eyes, hair, and central nervous system (CNS). This condition occurs in less than 3% of the male population, with survival attributed to one of three potential mechanisms that prevent embryonic lethality: somatic mosaicism, where a post-zygotic mutation allows partial NF-κB expression in some cells, Klinefelter’s syndrome, which provides an extra X chromosome that may carry a functional gene copy, or hypomorphic mutations, which cause a milder NF-κB impairment [2].
Case presentation
A seven-month-old male infant presented with small, red, blistering lesions that had been present since birth. These lesions progressed to warty plaques and eventually healed into stripe-like hyperpigmented lines along the trunk and extremities, with mild involvement of the face and ears. The mother denied any family history of similar rashes. The infant exhibited no signs of neurological issues or a history of seizures and had met all developmental milestones to date. Dental abnormalities were not assessed, as he had not yet developed teeth.
On physical examination, hyperpigmented streaks and whorls were observed in a blaschkoid distribution on the bilateral ears, neck, trunk, upper extremities, and lower extremities suggestive of stage three IP (Figures 1, 2). The parents were counseled on the inheritance pattern and potential associations of IP, including developmental delays, seizures, and dental abnormalities. They were advised to continue regular well-child visits and seek a pediatric ophthalmology evaluation to rule out retinal vascular involvement.
Stage three of incontinentia pigmenti characterized by a blaschkoid distribution of hyperpigmented streaks and whorls on the bilateral ears, neck, trunk, upper extremities, and lower extremities
Hyperpigmented linear to reticulated streaks following Blaschko’s lines, observed on the left side of the infant, characteristic of the third stage of incontinentia pigmenti
At a four-month follow-up, an ophthalmologic evaluation found no retinal vascular involvement. Genetic testing revealed a normal karyotype with no IKBKG mutation, suggesting a somatic mosaicism variant. This aligns with previous findings, where 19 out of 28 reported cases of male patients with IP showed no detectable IKBKG mutation in blood, likely due to tissue-specific mosaicism [3]. However, despite the genetic findings, the clinical course of skin lesion development strongly supports IP. Given the potential morbidity associated with ocular and CNS involvement, close multidisciplinary follow-up is warranted.
Discussion
The cutaneous presentation of IP progresses through four distinct stages: inflammatory vesicular (papules, vesicles, and eosinophilic pustules), verrucous (warty plaques), hyperpigmented (linear or whorled brown pigmented lesions), and hypopigmented (atrophic lesions), typically following Blaschko’s lines. Early vesiculopustular lesions resolve through keratinocyte apoptosis, allowing replacement by normal genotype cells and facilitating progression through these stages [4]. Consequently, the hyperpigmentation observed in our patient, indicative of stage 3 IP, is expected to gradually fade with age and transition into linear areas of hypopigmentation.
With no familial history of IP, diagnosis requires the presence of at least one major criterion, such as neonatal vesicular rash, hyperpigmentation, or linear atrophic lesions [5]. Our patient met two major criteria: a neonatal vesicular rash reported by the parent and current hyperpigmentation. No minor criteria have been observed yet, though they may develop over time. The prevalence of these minor criteria sequelae is as follows: dental (80%), hair (28-38%), nail (40%), ocular (25-77%), and neurological (33%) [1].
Genetic testing confirmed that our patient has a normal karyotype (46, XY), suggesting that survival is due to somatic mosaicism. Somatic mosaicism arises from a postzygotic mutation that partially impairs NF-κB function, allowing a subset of cells to retain normal function and sustain viability [2]. The clinical manifestations of IP in male patients are highly variable, likely reflecting the underlying mosaicism. For example, Gupta et al. described a 5-year-old male patient who developed vesicular lesions that progressed to hyperpigmentation, along with hypodontia, intellectual delay, and primary nocturnal enuresis [6]. A case reported by Kenwrick et al. involved a male patient with severe hypodontia, sparse hair, and vision loss due to spontaneous vitreous hemorrhage. In contrast, other cases exhibited no systemic involvement, with affected male patients presenting only with IP-consistent skin findings [7]. These cases demonstrate varying degrees of cutaneous and systemic involvement, reinforcing the broad clinical spectrum of IP in the male population.
The variability in disease severity in the male population with suspected somatic mosaicism raises the broader question of whether sex influences the overall disease burden in IP. Pacheco et al. found that in eight of nine male patients with a normal karyotype and no familial history of IP, lesions were confined to a single extremity, unlike female patients who exhibited a broader distribution along Blaschko’s lines [8]. However, this pattern was not observed in our patient, further underscoring the variability in cutaneous involvement. This difference may be attributed to the timing and extent of the postzygotic mutation leading to somatic mosaicism. While cutaneous patterns in males often differ from those in females, Minić et al. found no significant differences in the prevalence of CNS abnormalities between sexes in a study of 795 IP patients (719 female patients and 76 male patients), suggesting that other extracutaneous symptoms may follow similar trends [9]. Although genetic factors affect survival rates between sexes, males do not necessarily have more severe systemic involvement. Both sexes can develop complications with varying severity, emphasizing the need for further research to understand sex-based differences in the clinical presentation of IP.
While somatic mosaicism is the most common survival mechanism in males, Klinefelter syndrome (47, XXY) also enables survival by creating a heterozygous genotype in which X-inactivation favors the wild-type IKBKG allele [2]. Additionally, hypomorphic mutations arising in early embryonic development can result in a less deleterious form of IKBKG, providing another segue for survival. However, whereas somatic mosaicism and Klinefelter syndrome do not significantly impact disease severity based on sex, hypomorphic mutations produce a distinct phenotype. Females typically present with milder symptoms, often asymptomatic or limited to cutaneous manifestations, while males deviate from the typical IP phenotype and exhibit hypohidrotic ectodermal dysplasia and severe immunodeficiency [7,10].
Conclusions
This case demonstrates a male infant with IP, illustrating the rarity of survival of male infants and highlighting somatic mosaicism as a potential mechanism. While cutaneous progression is crucial for diagnosis, its severity does not necessarily predict systemic involvement. Given the risks of neurological and ophthalmological complications, multidisciplinary care and close long-term follow-up are essential for optimal clinical outcomes. The absence of photographic documentation of stages 1 and 2, along with the lack of IKBKG mutation detection, presents limitations that could have strengthened diagnostic certainty. Despite these constraints, this case contributes to the growing understanding of IP in male patients and underscores the importance of ongoing clinical observation and research
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Incontinentia pigmenti (Bloch-Sulzberger syndrome): a systemic disorder Cutis Ehrenreich M Tarlow MM Godlewska-Janusz E Schwartz RA 355362792007 https://pubmed.ncbi.nlm.nih.gov/17569396/17569396 · pubmed ↗
- 2A case of a surviving male infant with incontinentia pigmenti Ann Dermatol Song JY Na CH Chung BS Choi KC Shin BS 1341372020082730317710.5021/ad.2008.20.3.134PMC 4903964 · doi ↗ · pubmed ↗
- 3Diagnostic and molecular genetic challenges in male incontinentia pigmenti: a case report Acta Derm Venereol Gregersen PA Sommerlund M Ramsing M Gjørup H Rasmussen AA Aggerholm A 7417429320132357211610.2340/00015555-1593 · doi ↗ · pubmed ↗
- 4Uncovering incontinentia pigmenti: from DNA sequence to pathophysiology Front Pediatr How KN Leong HJ Pramono ZA Leong KF Lai ZW Yap WH 9006061020223614782010.3389/fped.2022.900606 PMC 9485571 · doi ↗ · pubmed ↗
- 5Incontinentia pigmenti An Bras Dermatol Poziomczyk CS Recuero JK Bringhenti L 26368920142462664510.1590/abd 1806-4841.20142584 PMC 3938351 · doi ↗ · pubmed ↗
- 6Case reports of incontinentia pigmenti in males Indian J Dermatol Gupta KD Padhiar BB Karia UK Shah BJ 32858201310.4103/0019-5154.113998 PMC 372691123919034 · doi ↗ · pubmed ↗
- 7Survival of male patients with incontinentia pigmenti carrying a lethal mutation can be explained by somatic mosaicism or Klinefelter syndrome Am J Hum Genet Kenwrick S Woffendin H Jakins T 121012176920011167382110.1086/324591 PMC 1235532 · doi ↗ · pubmed ↗
- 8Incontinentia pigmenti in male patients J Am Acad Dermatol Pacheco TR Levy M Collyer JC 2512555520061684450710.1016/j.jaad.2005.12.015 · doi ↗ · pubmed ↗