Genomic insights into host–Endozoicomonadaceae cophylogeny
Zhuang Shao, Jian Zhang, Jiaxin Li, Jie Li

TL;DR
This study explores how the genomes of Endozoicomonadaceae bacteria influence their long-term relationships with their hosts.
Contribution
The study introduces a genomic approach to analyze cophylogeny, linking genomic erosion and functional genes to host associations.
Findings
Endozoicomonadaceae show a cophylogenetic pattern with their hosts.
Smaller, more eroded genomes correlate with weaker host associations.
Specific genes related to infection and symbiosis are found in closely associated Endozoicomonadaceae.
Abstract
The congruence between host and symbiont phylogenies reflects the evolutionary links among ecologically important interactions. As potential key symbionts, the members affiliated to the family Endozoicomonadaceae have previously been investigated for the cophylogenetic relationship with their hosts using their 16S rRNA gene sequences. However, this approach neglects the genomic features of symbionts that may influence the long-term associations between Endozoicomonadaceae members and their hosts. Here, we collected available high-quality genomes of Endozoicomonadaceae from diverse hosts and investigated their genomic features, including genome size, phages, insertion elements and the composition of functional genes. We also tested the host–Endozoicomonadaceae cophylogeny and examined the correlation between the cophylogenetic squared residuals and the genomic features of…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
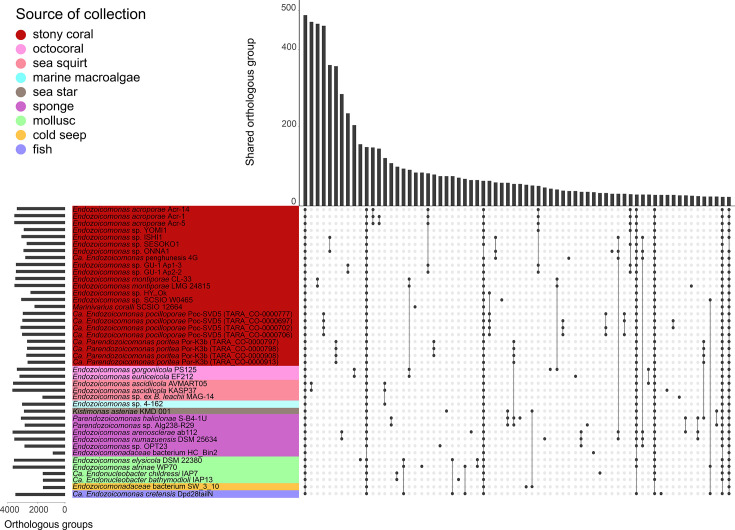 Fig. 1
Fig. 1 Fig. 2
Fig. 2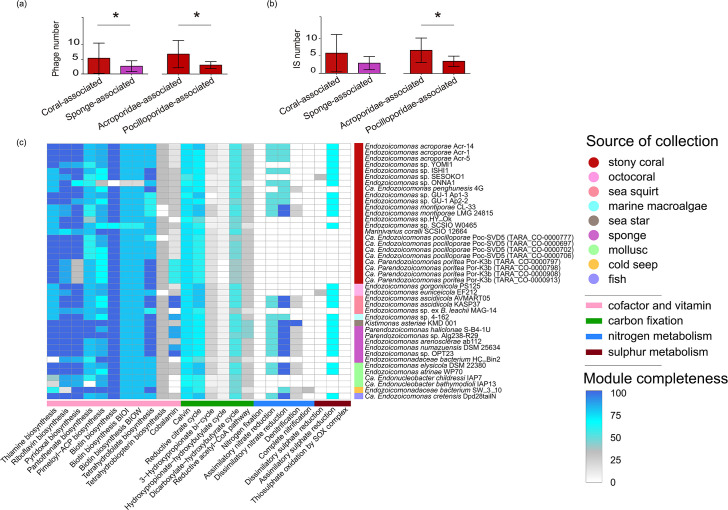 Fig. 3
Fig. 3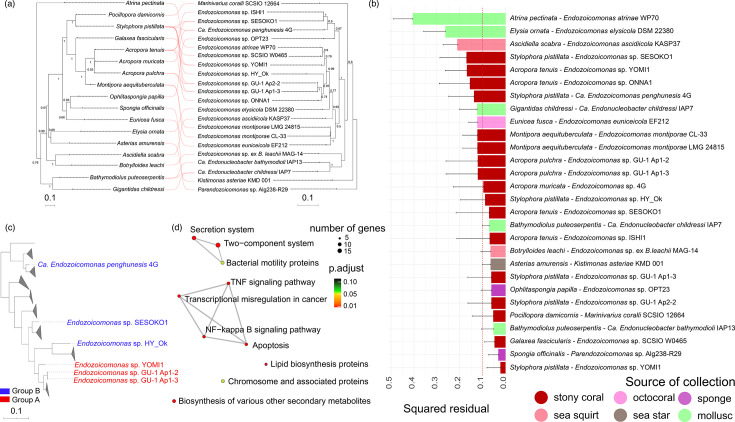 Fig. 4
Fig. 4- —http://dx.doi.org/10.13039/501100001809 National Natural Science Foundation of China
- —http://dx.doi.org/10.13039/501100001809 National Natural Science Foundation of China
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Phylogenetic Studies · Parasitic Infections and Diagnostics · Metal Extraction and Bioleaching
Data Summary
All supporting data have been provided within the article or through supplementary data files.
Introduction
The symbiotic relationship between microbes and marine animals is fundamental to the survival of marine organisms in various environments and underpins the health of marine ecosystems [1]. Among the diverse array of marine host-associated bacteria, the family Endozoicomonadaceae is notable for its abundance, commonness and functional diversity. Because of these features, it is regarded as a key group of bacterial symbionts in marine ecosystems [23]. The family Endozoicomonadaceae (Gammaproteobacteria, Oceanospirillales) [4] includes the genera Kistimonas [5], Parendozoicomonas [4], Sansalvadorimonas [6] and Endozoicomonas [7]. These bacteria inhabit a wide range of marine environments and various marine animals [8]. They are common bacterial symbionts of diverse invertebrates, such as stony corals, sponges, octocorals, sea anemones and sea squirts [9]. But there are also Endozoicomonadaceae species in seabream and deep-sea molluscs that were defined as pathogens [1011]. Recent comparative genomic analyses revealed variability in the size of Endozoicomonadaceae genomes, which tend to be relatively large and exhibit varying degrees of erosion caused by phages and insertion elements [1213]. Several studies have indicated that bacterial genome size can reflect the extent of its specialized symbiosis with the host [1415]. Additionally, diverse insertion elements may help bacteria adapt to their hosts [16], potentially influencing gene functions or the evolution of bacteria [1718]. Moreover, Endozoicomonadaceae species also harbour many genes related to symbiosis, such as the type III secretion system, cobalamin biosynthesis and dimethylsulfoniopropionate metabolism. These gene functions contribute to the host*–Endozoicomonadaceae* associations [121619]. Understanding the long-term, intimate relationships between symbionts and hosts is crucial for unravelling the ecological and evolutionary dynamics shaping host–symbiont interactions [2021]. However, it remains uncertain to what extent the genomic features of Endozoicomonadaceae members reflect long-term associations with their hosts.
In this context, investigating the cophylogenetic pattern between members of Endozoicomonadaceae and their hosts becomes imperative. Understanding the evolutionary histories and coevolutionary dynamics between these symbiotic partners would provide insights into their interactions [22]. Previous studies have hinted at a cophylogenetic pattern between Endozoicomonadaceae members and their stony coral and sponge hosts based on 16S rRNA genes [2324]. The limitations of using this marker gene may hinder a comprehensive understanding of the underlying mechanisms driving host–symbiont associations. Therefore, a genomic perspective is essential for elucidating the intricate relationships between Endozoicomonadaceae members and their hosts, especially given their ecological significance and wide distribution across marine ecosystems.
In this work, we collected nearly complete genomes of Endozoicomonadaceae members associated with marine invertebrates, fish, macroalgae and cold-seep environments, analysed the basic structure and functional features of these genomes and investigated the cophylogenetic pattern between Endozoicomonadaceae members and their hosts. We further explored the potential associations of Endozoicomonadaceae genome structure and gene functions with host*–Endozoicomonadaceae* cophylogenetic squared residuals. This study aims to investigate the genomic basis of host*–Endozoicomonadaceae* symbiosis and its implications for ecological and evolutionary dynamics.
Methods
Endozoicomonadaceae genome collections
The genomes (including cultivated genomes, single-cell amplified genomes and metagenomic assembled genomes) of the Endozoicomonadaceae members were collected from the National Center for Biotechnology Information (NCBI) and Zenodo (https://doi.org/10.5281/zenodo.7840163). These genomes were reannotated using Prokka v1.14.6 [25] to avoid the incongruence of different annotation schemes. They were checked for completeness, contamination, heterogeneity and other basic genome information with CheckM v1.0.12 [26]. Following the quality check, 42 Endozoicomonadaceae genomes were selected for this study. Thirty-eight of these genomes from shallow-sea hosts exhibited completeness ranging from 90 to 99.6% and contamination levels ranging from 0 to 5.2%. Additionally, four genomes from deep-sea invertebrate hosts and environmental samples showed completeness ranging from 73.8 to 88.1% and contamination levels ranging from 0 to 2.5%. Due to the rarity and difficulty of collecting these deep-sea samples, they were not filtered based on quality metrics (Table S1, available in the online Supplementary Material).
Identification of genomic features
All orthologous groups of the genomes were identified using OrthoFinder v2.5.5 [27], and the visualization of orthologous groups in each genome was performed via UpSetR [28]. The whole-genome average nucleotide identity (ANI) was calculated using FastANI v1.32 [29]. Domain information, COG and KEGG orthology were annotated using eggNOG-mapper v2.1.12 [30]. Furthermore, EnrichM v0.6.5 (https://github.com/geronimp/enrichM) was used to reveal the complete KEGG orthology modules in the genomes. Enrichment analysis was used to map genes to pathways, and a network plot was generated to display shared genes between pathways, both using the clusterProfiler package [31]. The Phage Search Tool Enhanced Release [32] was used to detect the phage regions and identify the possible phage species. UGENE [33] was used to search for repeat sequences from genomes with a minimum identity of 98% and a minimum length threshold of 500 bp (rRNA was not included) [16]. The ISfinder [34] was used to further detect the insertion elements in the repeat sequences with the 10^−6^ e-value threshold [16]. The geNomad v1.7.4 [35] with the conservative filter was used to identify and annotate all phage sequences integrated into the genomes of Endozoicomonadaceae species. We performed two-tailed t-tests on the number of insertion elements and phages in the Endozoicomonadaceae genomes collected from stony corals and sponges. Similar t-tests were also conducted on Endozoicomonadaceae genomes collected from the families Acroporidae and Pocilloporidae. These groups were selected for t-tests based on the sufficient sample numbers required for the analysis.
Endozoicomonadaceae and host phylogenetic reconstructions
Single-copy orthologous genes of Endozoicomonadaceae genomes were extracted from the OrthoFinder results. Each gene was aligned using muscle v5 [36], and low-quality alignments were removed using Gblocks v0.91b [37]. ModelFinder [38] was used to select the best evolutionary model. MrBayes v3.2 [39] was used for phylogenetic analysis with the WAG+G4 model, applying the Markov Chain Monte Carlo method using two independent runs of 2 000 000 generations, sampling at every generation with an initial burn-in of 1000 generations. Hahella chejuensis KCTC 2396 (Oceanospirillales, Hahellaceae) was selected as the outgroup. In the host phylogenetic analysis, hosts with clear species information were selected for phylogenetic reconstruction (except for the host Diplodus puntazzo, which was infected with Endozoicomonadaceae species under experimental conditions). Tridacna crocea was selected as the outgroup. For the selection of the host marker gene, representative sequences for 18S rRNA, cytochrome c oxidase subunit I and cytochrome b were collected from NCBI. These sequences were processed using the steps (muscle, Gblocks, ModelFinder and MrBayes with the GTR+G+F model) as those used for the phylogenetic reconstruction of Endozoicomonadaceae members.
Cophylogenetic analysis
To test for phylogenetic congruence, the phylogenetic trees of the host and Endozoicomonadaceae were assessed using the Procrustean Approach to Cophylogeny (PACo) [40] with 100 000 permutations. This distance-based global fit method quantifies the topological congruence between the two phylogenetic trees. A P-value less than 0.01 indicates a strong, non-random association between the two phylogenies. Additionally, the squared residuals quantify the extent to which symbiont species track their hosts. A smaller squared residual indicates a closer cophylogenetic relationship between the host and symbiont. To improve the accuracy of the cophylogenetic analysis, additional host species of Endozoicomonadaceae were collected to identify other hosts in which these bacteria were found, beyond the ones from which they were originally isolated. We extracted 16S rRNA gene sequences from Endozoicomonadaceae genomes and performed a blastn analysis in NCBI. If the 16S rRNA gene sequences from Endozoicomonadaceae genomes exhibited more than 99% identity with other 16S gene sequences of Endozoicomonadaceae species in the database, we collected the corresponding host species information. A total of 22 Endozoicomonadaceae members with identified host species were subjected to cophylogenetic analysis (Table S1).
The software eMPRess [41] was used to assess the cophylogenetic events between Endozoicomonadaceae members and their hosts. Cophylogenetic events, including cospeciations, duplications, transfers and losses, were statistically tested through 100 generations of randomization, with a P-value lower than 0.01 indicating that the events were unlikely to have occurred by chance.
The cophylogenetic analysis was conducted at a bacterial family level, as higher taxonomic ranks are less likely to have evolved with their hosts, whilst lower taxonomic ranks may introduce more noise [23]. We also tested the correlation between the host*–Endozoicomonadaceae* cophylogenetic jackknifed squared residuals and the genome size, number of phages and total insertion elements using the Spearman method in RStudio [42].
Results
Genome features and phylogenomic analysis of the family Endozoicomonadaceae
This study encompassed a total of 42 genomes from Endozoicomonadaceae members, with genome sizes ranging from 2.00 to 6.98 Mb (calculated based on 100% completeness; Table S1). The largest genome, Endozoicomonas sp. SCSIO W0465, contained 6571 genes, whilst the smallest genome, Endozoicomonadaceae bacterium HC_Bin2, contained 1402 genes. In total, 190 375 genes were annotated across the 42 genomes. Approximately 2% of these genes were identified as single-copy orthologous groups shared by all genomes. A total of 94.9% of these genes were grouped into 14 567 orthologous clusters. Moreover, we found that two sea squirt-associated Endozoicomonas ascidiicola strains, AVMART05 and KASP37, shared 469 orthologous groups with a genome similarity of 98.77%. Similarly, the two stony coral-associated Endozoicomonas montiporae strains, CL-33 and LMG 24815, shared 464 orthologous groups with a genome similarity of 99.88% (Table S2). Endozoicomonadaceae members from each source of collection showed unique orthologous groups, except for uncultured Endozoicomonas sp. 4–162 collected from the marine macroalgae (Fig. 1).
The number of orthologous groups in Endozoicomonadaceae genomes. The bar plot on the left indicates the number of orthologous groups in each genome. The bar plot on the top indicates the number of orthologous groups shared among the genomes. The dots and lines indicate the genomes from which the shared orthologous groups originate. The colours represent genomes collected from different sources.
For the phylogenomic analysis, the phylogenetic tree of the members of Endozoicomonadaceae was constructed based on 243 single-copy marker genes. Parendozoicomonas members collected from the same type of host were observed to cluster together. Two unclassified strains, Endozoicomonadaceae bacterium SW_3_10 and Endozoicomonadaceae bacterium HC_Bin2, clustered separately with Kistimonas asteriae KMD 001 and ‘Marinivarius coralli’ SCSIO 12664, respectively. Most Endozoicomonas members collected from the same type of host were observed to cluster together, although those associated with stony corals formed three distinct groups (Fig. 2).
Phylogenomic analysis of the members of the Endozoicomonadaceae family. The evolutionary history was inferred using a Bayesian method based on 243 single-copy marker genes. The genome of H. chejuensis KCTC 2396 was used as the outgroup to root the tree. The coloured bars next to the tree display genus information of the genomes.
Insertion elements in genomes of Endozoicomonadaceae
The number of insertion elements in Endozoicomonadaceae genomes ranged from 0 to 22, involving 13 distinct insertion element families (Table S1). No insertion elements were observed in Endozoicomonadaceae bacterium SW_3_10 collected from cold-seep seawater or in Endozoicomonas sp. OPT23 collected from Ophlitaspongia papilla, which may be attributed to their incomplete genomes (88 and 99% completeness, respectively). In the unpaired t-test with Welch’s correction (Fig. 3a and b), there was no significant difference in the number of insertion elements between stony coral-associated and sponge-associated Endozoicomonadaceae genomes (t=2.059, P=0.0505). However, a significant difference was observed in the number of insertion elements between Endozoicomonadaceae genomes associated with the families Acroporidae and Pocilloporidae (t=2.798, P=0.0151).
Differences in the number of phages and insertion elements, as well as in the metabolic features of Endozoicomonadaceae genomes. (a) T-tests comparing the number of phages in Endozoicomonadaceae genomes from different host sources. (b) T-tests comparing the number of insertion elements in Endozoicomonadaceae genomes from different host sources. (c) Major functional modules of Endozoicomonadaceae genomes. The colour gradient from white over grey to blue reflects an increase in the completeness of KEGG modules related to each function.
As a type of insertion element, the phage analysis revealed that a total of 2739 phage genes were annotated in all Endozoicomonadaceae genomes. Most phage species infecting Endozoicomonadaceae belong to the order Caudoviricetes, including Autographiviridae, Ackermannviridae, Demerecviridae and Pedosvirus (Table S3). By performing functional annotation of phage genes with Pfam, COG and KEGG databases, most of these genes encode phage proteins, such as phage major capsid protein E, phage tail tape-measure proteins and bacteriophage lambda head decoration protein D. However, we also found the vitamin B12 synthesis protein (cobalamin biosynthesis protein CobT) in phage sequences from Endozoicomonas gorgoniicola PS125 and Endozoicomonas atrinae WP70 (Table S3). Furthermore, different numbers of phages were observed between stony coral-associated and sponge-associated Endozoicomonadaceae genomes (t=2.102, P=0.046). A total of 106 phage regions were identified in the investigated Endozoicomonadaceae genomes collected from stony corals. Among these, 17 phages were intact, and PHAGE_Pseudo_phi3_NC_030940 was detected in eight genomes (Table S4). Additionally, a total of 14 phages were identified in the Endozoicomonadaceae genomes collected from sponges. Moreover, the number of phages was significantly different in the genomes of Endozoicomonadaceae members associated with Acroporidae and Pocilloporidae (t=2.658, P=0.0179).
Metabolic potential of the Endozoicomonadaceae family
Through the annotation of gene functions in Endozoicomonadaceae genomes, eukaryotic repeat proteins (ankyrin repeats, WD40 repeats, tetratricopeptide repeat and leucine-rich repeat protein) were found in most (93%) of the studied Endozoicomonadaceae genomes. Genes of the type III secretion system were ubiquitous in all investigated Endozoicomonadaceae genomes (Table S5). The completeness of KEGG modules for putative gene functions of Endozoicomonadaceae genomes was assessed (Fig. 3c). In the cofactor and vitamin metabolism pathways, thiamine biosynthesis, riboflavin biosynthesis, pyridoxal biosynthesis and biotin biosynthesis were intact in most Endozoicomonadaceae genomes. K. asteriae KMD 001 and Parendozoicomonas sp. Alg238-R2 both exhibited seven complete cofactor and vitamin biosynthesis modules, except the pathways for tetrahydrobiopterin and cobalamin biosynthesis. ONNA1, associated with stony coral, only possessed the complete riboflavin biosynthesis module. Endozoicomonas sp. ex B. leachii MAG-14, Ca. Endonucleobacter childressi IAP7 and Ca. Endonucleobacter bathymodioli IAP13 retained the complete biotin biosynthesis module. Endozoicomonadaceae bacterium HC_Bin2, associated with sponge, lacked complete genes involved in thiamine, riboflavin, pyridoxal and tetrahydrofolate biosynthesis, whilst other sponge-associated members potentially produced these vitamins with complete biosynthetic modules. Additionally, results of functional gene analysis suggested that Endozoicomonadaceae did not appear to be capable of carbon fixation, nitrogen fixation, sulphate reduction or thiosulphate oxidation. Most coral-associated Endozoicomonadaceae members lacked complete genes involved in the assimilatory and dissimilatory nitrate reduction, denitrification and nitrification. K. asteriae KMD 001, associated with the sea star host, had the complete denitrification pathway. Additionally, the genes nosZ, nosF and nosY, responsible for the reduction of N_2_O, were observed in the genome of K. asteriae KMD 001. Phosphatidylcholine is the major membrane-forming phospholipid in eukaryotes and may be present in about 15% of bacteria [43]. Only Endozoicomonas sp. HY_Ok and Ca. Endozoicomonas pocilloporae Poc-SVD5 (TARA_CO-0000777) showed a complete phosphatidylcholine biosynthesis pathway. Ectoine acts as a cellular defence to protect cells against stress [44]. Amongst the Endozoicomonadaceae genomes, five contained a complete ectoine biosynthesis pathway, whilst the remaining genomes contained partial pathways (Table S6).
Cophylogeny and correlation between cophylogenetic squared residuals and genomic features of the Endozoicomonadaceae family
Cophylogenetic analysis showed that the known members of the Endozoicomonadaceae family and their hosts had phylogenetic congruence (the observed m^2^ is 2.80251, P-value=4e-05<0.01), indicating that the phylogeny of Endozoicomonadaceae members was not randomly associated with the phylogeny of their hosts (Fig. 4a). In all 29 squared residuals, 15 residuals were lower than the median squared residual (Fig. 4b). The strongest cophylogenetic pattern (with the smallest squared residual) was observed between Stylophora pistillata and Endozoicomonas sp. YOMI1. The weakest cophylogenetic pattern was observed between Atrina pectinata and E. atrinae. The results of the Spearman correlation analysis suggested a potentially positive trend between the size of Endozoicomonadaceae genomes and the cophylogenetic squared residuals, although this trend did not reach statistical significance. Moreover, there appeared to be a negative trend between the number of insertion elements and phages in the genomes with the cophylogenetic squared residuals, though these relationships also lacked statistical significance (Fig. S1).
Cophylogenetic analysis of host–Endozoicomonadaceae and enrichment analysis of specific genes. (a) The tanglegram of host–Endozoicomonadaceae. (b) Contributions of individual host–Endozoicomonadaceae links to the procrustean fit. Jackknifed squared residuals (bars) and upper 95% CIs (error bars) resulting from applying PACo to the distances of Endozoicomonadaceae members. The colour of each bar indicates the source of collection. (c) The phylogeny of Endozoicomonas members. Members of Endozoicomonas collected from S. pistillata are categorized into groups A and B. (d) Enrichment analysis results of group A-specific genes for KEGG pathways. The top 10 pathways are displayed. The thickness of the lines among pathways indicates the number of shared genes. The sizes of the circles represent a scale, indicating the gene numbers in pathways.
We further investigated the possible functional associations in the cophylogenetic process between S. pistillata and its associated Endozoicomonas symbionts on the basis of the relatively sufficient genomes. We divided the Endozoicomonas members collected from S. pistillata into two groups based on their cophylogenetic squared residuals: a smaller residual group A (Endozoicomonas sp. GU-1 Ap1-2, Endozoicomonas sp. GU-1 Ap1-3 and Endozoicomonas sp. YOMI1) and a larger residual group B (Endozoicomonas sp. HY_Ok, Endozoicomonas sp. SESOKO1 and Ca. Endozoicomonas penghunesis 4G) (Fig. 4b and c). In comparison of all the genes in group A with group B, we found that group A carried a total of 124 specific genes (Table S7). Enrichment analysis identified the top 10 pathways associated with these specific genes, involving the two-component system, secretion system, bacterial motility, lipid biosynthesis proteins, chromosome and associated proteins, biosynthesis of various other secondary metabolites and four pathways related to inflammation and immunity [45] (Fig. 4d).
The event-based cophylogenetic analysis using the eMPRess software revealed that host*–Endozoicomonadaceae* associations were involved in a total of 65 events, including 8 cospeciations, 9 duplications, 1 transfer and 47 loss events. The P-value from the randomization tests was less than 0.01 under cost region A (Fig. S2), indicating that the results were statistically significant and refuted the null hypothesis that the observed evolutionary events occurred by chance.
Discussion
In this study, we gathered multiple Endozoicomonadaceae genomes along with their host information. The distance-based cophylogenetic analysis provided strong evidence for a cophylogenetic pattern between Endozoicomonadaceae members and their hosts. To further explore evolutionary events during the cophylogenetic process, we employed the event-based cophylogenetic analysis, which revealed potential cospeciation events between Endozoicomonadaceae members and their hosts. Moreover, as the host*–Endozoicomonadaceae* cophylogenetic squared residuals decreased, the genome size of Endozoicomonadaceae members tended to decrease, whilst the number of phages and insertion elements appeared to increase, although these relationships lacked statistical significance. Furthermore, by focusing on S. pistillata, a host harbouring a relatively large number of Endozoicomonadaceae members, we identified that Endozoicomonadaceae members with closer associations with their hosts carried specific genes associated with infection processes and host–symbiont interactions. This is the first study to investigate host*–Endozoicomonadaceae* cophylogeny from a genomic perspective.
Previous studies have shown that some Endozoicomonadaceae genomes carry a large number of symbiosis-related genes, including those involved in host adaptation and synthesis of B vitamins [34647], and our analysis supports this finding. A large number of type III secretion system genes were identified in all investigated Endozoicomonadaceae genomes, and 39 genomes contained eukaryotic repeat proteins (Table S5). Our analysis also revealed that Endozoicomonadaceae genomes exhibited significant potential in synthesizing B vitamins, especially B7 (biotin), B6 (pyridoxine), B2 (riboflavin) and B1 (thiamine). Additionally, we identified some genes in Endozoicomonadaceae genomes that may play a role in host*–Endozoicomonadaceae* adaptation. Ectoine, initially discovered as an osmoprotectant in anaerobic photobiology [48], was shown to enhance protein stability and act as a whole-cell stabilizer against various environmental stressors such as heating, freezing and UV radiation [44]. Among the genomes of the Endozoicomonadaceae family, five genomes had an intact pathway for ectoine biosynthesis, whilst others possessed a partially intact pathway (Table S6), suggesting that Endozoicomonadaceae members may exhibit the ability to adapt to complex microenvironments. A recent study [43] demonstrated that bacterial phosphatidylcholine plays an important role in the binding of bacteria to host macrophages and in promoting motility, biofilm formation and colonization of some pathogens, which may contribute to interactions between bacteria and their eukaryotic hosts. Endozoicomonas sp. HY_Ok and Ca. E. pocilloporae Poc-SVD5 (TARA_CO-0000777) had a complete phosphatidylcholine biosynthesis pathway (Table S6). However, the role of phosphatidylcholine in host*–Endozoicomonadaceae* interactions remains unknown. Moreover, our analysis of phages and insertion elements in Endozoicomonadaceae genomes revealed varying degrees of genome erosion (Table S1). The expression of phage genes during infection may confer new functions on bacteria [1749]. We found that phages carrying the vitamin B12 synthesis protein (cobalamin biosynthesis protein CobT) were integrated into both E. gorgoniicola PS125 and E. atrinae WP70 (Table S3), suggesting that phage infections may facilitate functional associations between Endozoicomonadaceae members and their hosts.
Cophylogenetic patterns can reveal a prolonged evolutionary history of host–symbiont associations [50]. In our study, both distance-based and event-based analyses suggested that the evolutionary histories of the two pairwise comparisons (host*–Endozoicomonadaceae*) are not independent. The distance-based cophylogenetic analysis indicated significant phylogenetic congruence between the recognized Endozoicomonadaceae members and their hosts (Fig. 4a). Additionally, the notably small cophylogenetic squared residual observed between S. pistillata and Endozoicomonas sp. YOMI1 may underscore their close evolutionary association. The event-based cophylogenetic analysis identified an 8 out of 65 cospeciation rate, potentially contributing to the observed phylogenetic congruence. Moreover, we also found that the transfer event constituted only 1 out of 65 of all events, suggesting that members of the Endozoicomonadaceae family may rarely switch hosts. Additionally, the low transfer rate may also be attributed to the high cost of host transfer events, which are often replaced by duplicates and losses [51]. Whilst it is in our knowledge that a time-calibrated phylogeny may produce more accurate results when testing for cophylogenetic events [2351], eMPRess, a recognized testing software, currently does not support this need. As a result, the timing of cophylogenetic events occurring between the members of the Endozoicomonadaceae family and their hosts cannot be accurately determined. Furthermore, when evaluating host–symbiont cophylogenetic events, the limited availability of symbiont data can result in incomplete phylogenetic branches for symbionts. This limitation may increase the occurrence of lost events [50] and reduce the proportion of evolutionary events favouring host–symbiont cophylogeny. A more comprehensive exploration of the host*–Endozoicomonadaceae* cophylogenetic relationship will require a more accurate and detailed analysis of a larger dataset.
The dependence of symbiont phylogeny on host phylogeny is a basic principle for testing cophylogeny [51], whilst the genome size of bacterial symbionts can reflect their evolutionary dependency on the host when they are engaged in obligate symbiosis, with smaller genomes indicating a more specialized symbiotic relationship [14]. Consistent with previous reports, the size of genomes varied widely among members of the Endozoicomonadaceae family investigated in this study, indicating the possibility of a facultative to obligate symbiotic life stage within the family of Endozoicomonadaceae. Moreover, a potential trend of decreased cophylogenetic squared residuals between Endozoicomonadaceae members and their hosts as the Endozoicomonadaceae genomes shrank, although this trend was not statistically significant (Fig. S1A), suggesting that Endozoicomonadaceae genome size may reflect the host*–Endozoicomonadaceae* cophylogenetic relationship, providing a new perspective for understanding the host–symbiont cophylogeny.
In addition to genome size, a variety of phages and insertion elements offer insights into the infection and colonization histories of various marine hosts, facilitated by frequent divergence occurrences [12]. Furthermore, we observed potential trends showing increased cophylogenetic squared residuals between Endozoicomonadaceae species and their hosts as the number of phages and insertion elements increased, though these relationships were not statistically significant (Fig. S1B and C). Infection of bacterial genomes by phages and insertion elements causes alterations in genome structure and promotes the horizontal transfer of genes [5253]. This process contributes to bacterial adaptation and evolution [54] and may also play a role in host–bacteria cophylogeny.
Bacteria that exhibit a cophylogenetic pattern with their hosts may be functionally interdependent with them, fostering stable symbiotic relationships over long evolutionary periods [5556]. In our analysis, Endozoicomonadaceae members in group A, which had stronger connections with the host S. pistillata than those in group B, carried specific genes related to the two-component system, bacterial motility proteins and immune response (Fig. 4d). These functions may play a role in processes including host infection and interaction [455758], contributing to the long-term symbiosis between hosts and Endozoicomonadaceae members. Therefore, grouping symbionts based on their phylogenetic positions and the cophylogenetic squared residuals of host–symbiont pairs may be an effective method for elucidating the long-term functional associations between hosts and symbionts.
In contrast to previous analyses of host*–Endozoicomonadaceae* cophylogeny based on Endozoicomonadaceae 16S rRNA genes [2324], our study firstly demonstrates the cophylogenetic pattern between Endozoicomonadaceae members and various marine hosts based on Endozoicomonadaceae genomes. Secondly, this study discusses the potential correlation between the genomic features of investigated Endozoicomonadaceae members, such as genome size, phages, insertion elements and gene functions, and their long-term symbiotic relationship with their hosts. We suggest that the genomic features of symbionts should be considered in future studies of host–symbiont cophylogeny.
supplementary material
10.1099/mgen.0.001384Uncited Supplementary Material 1.
10.1099/mgen.0.001384Uncited Supplementary Material 2.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Wilkins LG Leray M O’Dea A Yuen B Peixoto RS et al Host-associated microbiomes drive structure and function of marine ecosystems P Lo S Biol 201917 e 300053310.1371/journal.pbio.300053331710600 PMC 6874084 · doi ↗ · pubmed ↗
- 2Neave MJ Apprill A Ferrier-Pagès C Voolstra CR Diversity and function of prevalent symbiotic marine bacteria in the genus Endozoicomonas Appl Microbiol Biotechnol 20161008315832410.1007/s 00253-016-7777-027557714 PMC 5018254 · doi ↗ · pubmed ↗
- 3Hochart C Paoli L Ruscheweyh H-J Salazar G Boissin E et al Ecology of Endozoicomonadaceae in three coral genera across the Pacific Ocean Nat Commun 202314303710.1038/s 41467-023-38502-937264015 PMC 10235432 · doi ↗ · pubmed ↗
- 4Bartz J-O Blom J Busse H-J Mvie JB Hardt M et al Parendozoicomonas haliclonae gen. nov. sp. nov. isolated from a marine sponge of the genus Haliclona and description of the family Endozoicomonadaceae fam. nov. comprising the genera Endozoicomonas, Parendozoicomonas, and Kistimonas Syst Appl Microbiol 201841738410.1016/j.syapm.2017.11.00429398077 · doi ↗ · pubmed ↗
- 5Choi EJ Kwon HC Sohn YC Yang HO Kistimonas asteriae gen. nov., sp. nov., a gammaproteobacterium isolated from Asterias amurensis Int J Syst Evol Microbiol 20106093894310.1099/ijs.0.014282-019661507 · doi ↗ · pubmed ↗
- 6Goldberg SR Haltli BA Correa H Kerr RG Description of Sansalvadorimonas verongulae gen. nov., sp. nov., a gammaproteobacterium isolated from the marine sponge Verongula gigantea Int J Syst Evol Microbiol 2018682006201410.1099/ijsem.0.00278129688166 · doi ↗ · pubmed ↗
- 7Kurahashi M Yokota A Endozoicomonas elysicola gen. nov., sp. nov., a gamma-proteobacterium isolated from the sea slug Elysia ornata Syst Appl Microbiol 20073020220610.1016/j.syapm.2006.07.00316904280 · doi ↗ · pubmed ↗
- 8da Silva DMG Pedrosa FR Ângela Taipa M Costa R Keller-Costa T Widespread occurrence of chitinase-encoding genes suggests the Endozoicomonadaceae family as a key player in chitin processing in the marine benthos ISME Commun 2023310910.1038/s 43705-023-00316-737838809 PMC 10576748 · doi ↗ · pubmed ↗