Boron, Aluminum, and Gallium Fluorides as Catalysts for the Defluorofunctionalization of Electron-Deficient Arenes: The Role of NaBArF4 Promoters
Wenbang Yang, Andrew J. P. White, Mark R. Crimmin

TL;DR
This paper explores how boron, aluminum, and gallium fluorides can act as catalysts in chemical reactions involving electron-deficient arenes, with a focus on how a specific additive improves their performance.
Contribution
The novel contribution is the discovery that NaBArF4 enhances catalytic activity by interacting with metal fluorides through weak interactions.
Findings
Metal fluorides [{(ArNCMe)2CH}MF2] catalyze defluorofunctionalization of electron-deficient arenes via a metathesis mechanism.
Addition of NaBArF4 recovers catalytic activity by forming weak M–F---Na interactions and lowering reaction barriers.
DFT calculations show that NaBArF4 polarizes the M–F bond, facilitating hydrido fluoride formation and catalytic turnover.
Abstract
A series of boron, aluminum, and gallium difluoride complexes [{(ArNCMe)2CH}MF2] (M = B, Al, Ga) are reported as catalysts for the defluorofunctionalization of electron-deficient arenes. Thiodefluorination reactions between TMS–SPh and poly(fluorinated aromatics) proceed under forcing conditions. Evidence is presented for the fluoride entering the catalytic cycle through a metathesis reaction with TMS–SPh to form metal thiolate intermediates, e.g., [{(ArNCMe)2CH}MF(SPh)], which are then nucleophiles for addition to the aromatic substrate, likely through a concerted SNAr mechanism. Attempts to expand the scope of reactivity to include the hydrodefluorination of electron-deficient arenes met with limited success. Activity could, however, be recovered through the addition of NaBArF4 as a catalytic additive (ArF = 3,5-C6H3(CF3)2). NMR titrations suggest that NaBArF4 is capable of…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
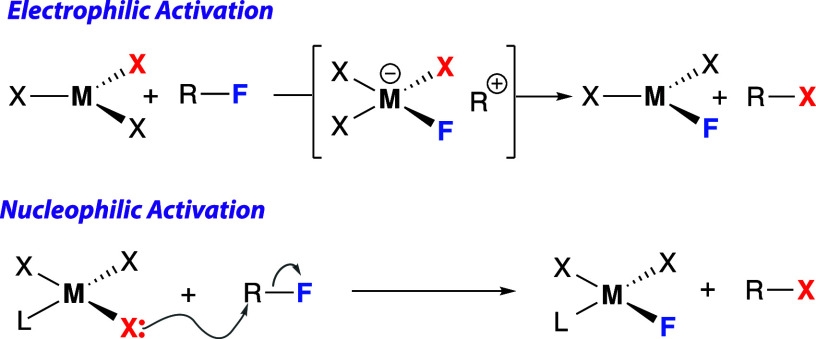 Figure 1
Figure 1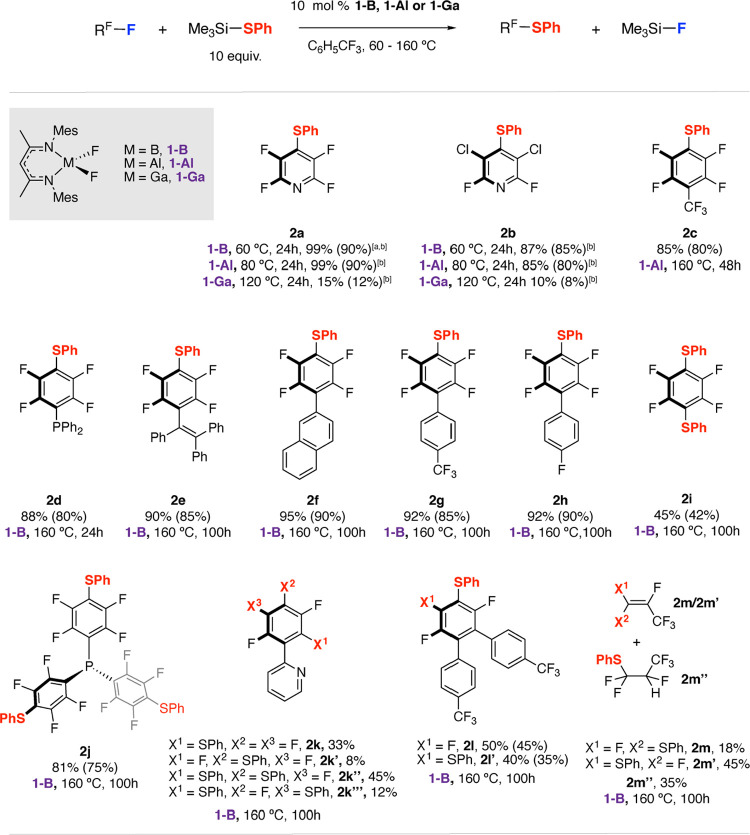 Figure 2
Figure 2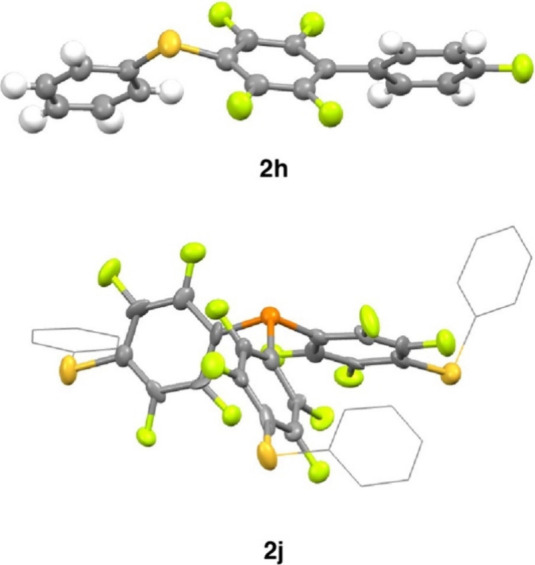 Figure 3
Figure 3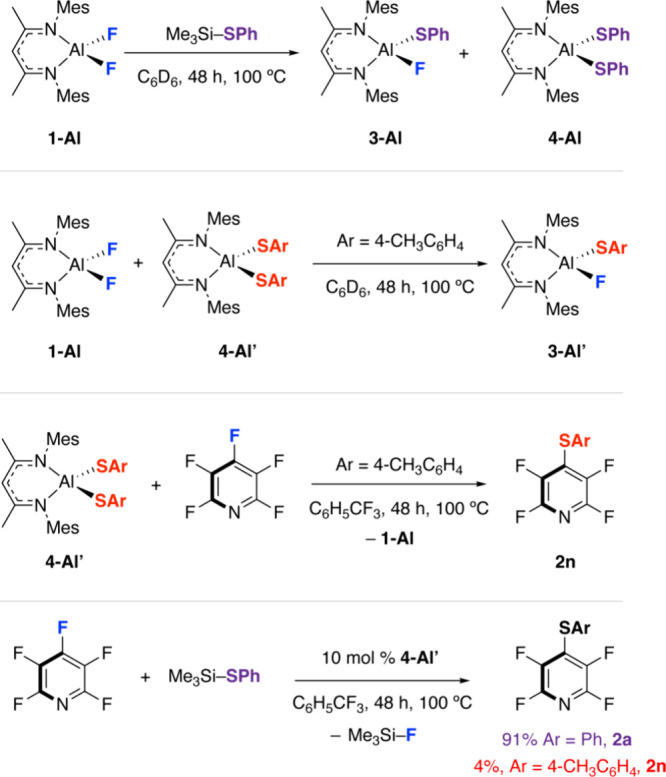 Scheme 1
Scheme 1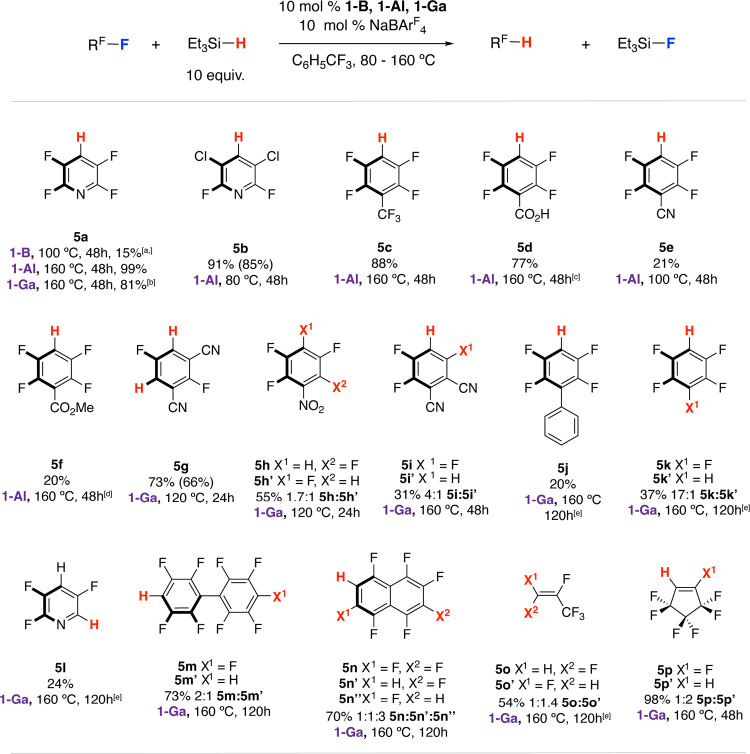 Figure 4
Figure 4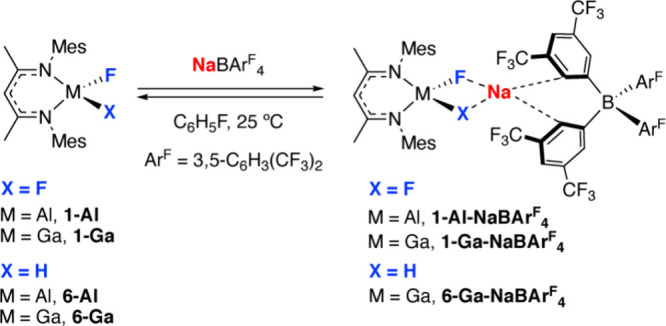 Scheme 2
Scheme 2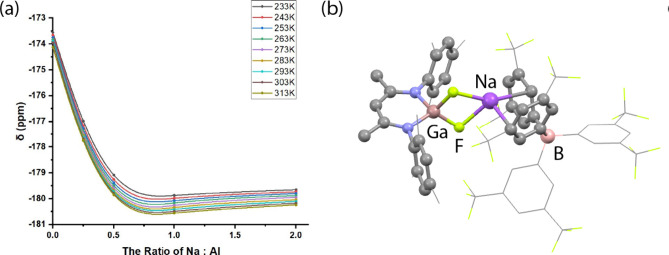 Figure 5
Figure 5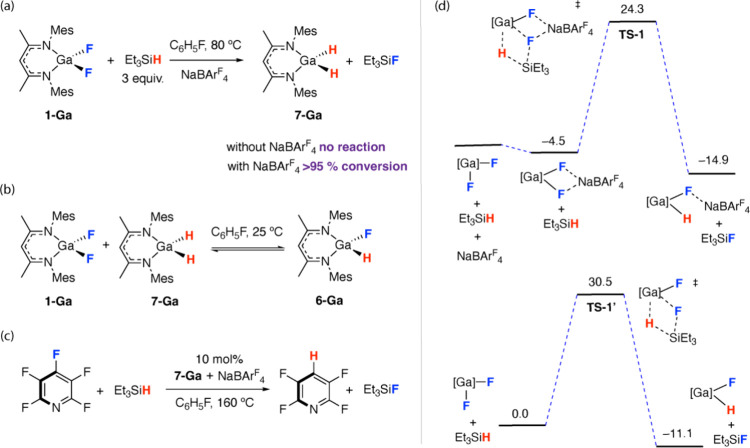 Figure 6
Figure 6- —H2020 European Research Council10.13039/100010663
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsFluorine in Organic Chemistry · Inorganic Fluorides and Related Compounds · Carbon dioxide utilization in catalysis
Introduction
Compounds of the group 13 elements boron, aluminum, and gallium are routinely employed as Lewis acids in catalysis. The high fluoride-ion affinities of three-coordinate group 13 compounds underpin numerous applications that break and functionalize strong carbon–fluorine bonds.^1^ For example, BF_3_–OEt_2_ and AlCl_3_ have been reported as catalysts for the Friedel–Crafts alkylation of arenes with fluoroalkanes.^2^ B(C_6_F_5_)3 has been shown to catalyze the hydrodefluorination of fluoroalkanes using Et_3_SiH as a terminal reductant.^3^ Similar reactivity has been achieved using aluminum chloride fluoride (ACF), a heterogeneous catalyst proposed to contain highly Lewis acid active sites based on aluminum.^4−8^ Aluminum compounds are also competent reagents^9,10^ and catalysts^11,12^ for carbon–heteroatom and carbon–carbon from fluoroalkanes. In the past few years, Frustrated Lewis Pair (FLP) catalysts based on group 13 compounds have been applied to highly selective catalytic transformations that allow the controlled functionalization of a single carbon–fluorine bond of sp^3^ CF_3_ or sp^2^ CF_2_H groups.^13−18^
In contrast, the use of nucleophilic group 13 compounds as the catalyst for carbon–fluorine bond functionalization is less common. It might be expected that by increasing the coordination number of a group 13 compound from three- to four- coordinate groups, its Lewis acid behavior could be tempered and the nucleophilic reactivity of the coordinated ligands could be exposed (Figure 1).^19^ For example, we can consider the complementary reactivity profiles of i-Bu_2_AlH and LiAlH_4_, with the latter hydride complex often considered to be the more nucleophilic reagent. The switch in electronic behavior might be expected to expand the scope of accessible substrates for catalytic transformations, as Lewis acid catalysts operate primarily on sp^3^ C–F bonds, whereas nucleophilic species would be capable of addition to sp^2^ C–F bonds. This hypothesis is largely untested. Examples of nucleophilic catalytic behavior are, however, more widespread for the transition metals.^20−22^
Selected reaction modes of group 13 compounds with carbon–fluorine bonds.
In the past 10 years, our research group have developed several new reactions that led to the generation of well-defined four-coordinate aluminum fluoride complexes as products.^23−25^ More recently, we became interested in the use of these complexes as potential catalysts for the functionalization of carbon–fluorine bonds. Here, we show that molecular fluoride complexes of boron, aluminum, and gallium are competent catalysts for the thiodefluorination and hydrodefluorination of electron-deficient arenes and alkenes. We report two defined approaches to catalysis: (i) an additive-free reaction and (ii) a catalytic protocol that relies on the use of NaBAr^F^4 (Ar^F^ = 3,5-C_6_H_3_(CF_3_)2) as an additive. Experimental and computational data are provided to support the formation of nucleophilic four-coordinate complexes as key intermediates and metal fluoride species as on-cycle species. Furthermore, we suggest that the additive plays a unique role in polarizing and activating metal–fluorine intermediates through the formation of weak M–F----Na interactions. Our findings complement existing uses of compounds based on main group elements such as tetrabutylammonium difluorophenylsilicate (TBAT), tetrabutylammonium fluoride (TBAF), or CsF as initiators for carbon–fluorine bond functionalization.^26^
Results and Discussion
Additive-Free Thiodefluorination of Arenes
A series of four-coordinate group 13 difluoride complexes 1-B, 1-Al, and 1-Ga supported by a sterically demanding β-diketiminate ligand were prepared. Initial experiments were conducted to screen a series of conditions for the reaction of pentafluoropyridine with group 13 catalysts using silanes as the terminal reductant. 1-B, 1-Al, and 1-Ga were found to be active catalysts for the thiodefluorination of electron-deficient arenes with silicon-based reagents at 10–20 mol % loading, between 60 and 160 °C, using α,α,α-trifluorotoluene as a solvent (Figure 2). Using Me_3_Si–SPh as a terminal reagent, we could selectively convert pentafluoropyridine into 2a. A control reaction showed a maximum of 10% conversion for this transformation after 24 h at 100 °C, when carried out in the absence of a catalyst. α,α,α-Trifluorotoluene was used as a solvent due to its high boiling point and its modest dielectric constant. Focusing on thiodefluorination, a scope of electron-deficient fluorinated aromatics was investigated. A series of substituted perfluoroarenes could be selectively converted to monosubstituted products 2b–2h. Under more forcing conditions, higher levels of substitution of pentafluoropyridine and perfluorotoluene could be achieved (see the Supporting Information for details). Hexafluorobenzene reacted selectively, giving 2i the product of 1,4-disubstitution. Similarly, P(C_6_F_5_)3 underwent a selective trisubstitution under catalytic conditions to yield 2j, which could be crystallographically characterized (Figure 3). 2j holds promise as a novel ligand for applications in catalysis. 2-(Perfluorophenyl)pyridine underwent nonselective thioylation to form isomers of 2k, primarily at the 2- and 4-position of the perfluorophenyl ring. This finding suggests that the pyridyl-directing group has little control on the reaction, arguing against the coordination of the substrate to the catalyst expected during Lewis acid catalysis. An electron-deficient polyfluorinated terphenyl underwent nonselective single and double substitution to form 2l and 2l′****. Hexafluoropropene is also reactive under catalytic conditions, providing a mixture of stereoisomers 2m:2m′ from thioylation of the terminal sp^2^C–F bonds along with a side product 2m″ derived from protonation, likely from adventitious water.
Scope of group 13 fluoride catalyzed defluorothioylation of fluorinated arenes and hexafluoropropene. aNMR yields were measured by 19F NMR spectroscopy using 1,2-difluorobenzene as an internal standard. Isolated yields in parentheses. bReactions run using 3 equiv of Me3Si–SPh.
Crystal structures of 2h and 2j. 50% probability ellipsoids, and selected hydrogen atoms are omitted for clarity.
For comparison, sodium thiolate anions are competent nucleophiles in S_N_Ar reactions with a range of fluoroarenes in polar solvents (e.g., ethylene glycol, pyridine, THF, DMF) under mild conditions.^27−30^ Similar reactions are reported to occur between fluoroalkenes or fluoroarenes and aryl thiols in the presence of K_2_CO_3_ as a base.^31,32^ Group 13 thiolate nucleophiles have also been reported for the defluorofunctionalization of alkyl fluorides.^9^ Catalytic approaches to thiodefluorination are less common but include fluoride metathesis reactions between thioesters and fluoroarenes catalyzed by either [RhH(PPh_3_)4] or 4-dimethylaminopyridine (DMAP).^33−35^
Qualitatively, for the synthesis of 2a and 2b, it was found that the catalysts were more active across the series 1-B > 1-Al > 1-Ga. The heavier group 13 catalysts require more forcing conditions and show more limited scope than the lightest boron analogue. Reaction trends are consistent with a nucleophilic mechanism with regioselectivity determined by the most electrophilic position of the aromatic ring. In contrast to known Lewis acid catalysts based on group 13, these reactions are selective for sp^2^C–F over sp^3^C–F bonds with CF_3_ groups being tolerated under catalytic conditions. Less electron-deficient arenes, i.e., those with lower fluorine content, do not react under optimized conditions.
To better understand the role of the catalyst, in two separate experiments, 1-B and 1-Al were reacted with 3 equiv of Me_3_Si–SPh. In the case of 1-B, no apparent reaction occurred,^36^ and 1-Al reacted with Me_3_Si–SPh after 48h at 100 °C in C_6_D_6_ to produce a 3:1 mixture of 3-Al and 4-Al along with Me_3_Si–F as a side product (Scheme 1). The latter was readily apparent from ^19^F NMR resonance at δ = −148.5 ppm. 4-Al could be crystallized from the mixture, and the structure was determined by single-crystal X-ray diffraction. It is likely that ligand exchange between fluoride and thiolate ligands is facile under the conditions of catalysis. A reaction between an independently prepared sample of the dithiolate complex 4-Al′ and 1-Al at 100 °C in C_6_D_6_ led to an intermolecular scrambling of these ligands with the formation of the cross-product 3-Al’, as evidenced by a broad singlet also found at δ = −148.5 ppm in the ^19^F NMR spectrum (Scheme 1). 4-Al′**** proved to be a competent nucleophile, as the reaction with pentafluoropyridine led to the formation of the defluorothioylation product 2n. 4-Al′**** was also shown to be catalytically active for the reaction of Me_3_Si–SPh with pentafluoropyridine under similar conditions as 1-Al, in this case, forming 2a as the major product along with small amounts of 2n derived from the catalytic loading of 4-Al′****.
Catalytic Relevance of Group 13 Thiolate ComplexesStoichiometric reaction between 1-Al and Me3Si–SPh. Reaction of an isolated dithiolate complex with 1-Al and pentafluoropyridine under stoichiometric and catalytic conditions.
NaBArF4 Promoted Hydrodefluorination of
Arenes
Attempts to extend the catalytic methodology to the hydrodefluorination of arenes met with limited success. 1-B, 1-Al, and 1-Ga were not active catalysts for the reaction of Et_3_SiH with pentafluoropyridine under catalytic conditions established for Me_3_Si–SPh. Following an extensive screen of conditions and additives, it was found that 10 mol % NaBAr^F^ could be used as an additive to effectively promote the reaction. Several silanes, including Et_3_SiH, Ph_2_SiH_2_, PhSiH_3_, Me_2_PhSiH, (MeO)_3_SiH, and (Me_2_SiH)_2_O, could be used as effective hydride sources with the reaction occurring in noncoordinating solvents, such as fluorobenzene or α,α,α-trifluorotoluene. α,α,α-Trifluorotoluene and fluorobenzene were used as solvents due to their high boiling points, modest dielectrics, and a known ability to solvate charge-separated species.
Under optimized conditions, pentafluoropyridine could be converted into 2,3,5,6-tetrafluoropyridine 5a in 99% using 10 mol % 1-Al + 10 mol % NaBAr^F^4 as a precatalyst mixture and 10 equiv of Et_3_SiH as the terminal reductant. Control reactions using solely 1-Al or NaBAr^F^4 led to minimal conversion; similarly, NaBPh_4_ was not an active additive under these conditions. Qualitatively, 1-Al and 1-Ga appeared to be more active catalysts than 1-B. The scope of hydrodefluorination was investigated. A series of electron-deficient aromatics, including nitro, nitrile, ester, and acid functional groups, underwent hydrodefluorination (5b–5n) with more forcing conditions and higher loadings required for less electron-deficient substrates (e.g., 5j,k, 5m,n). Fluorinated alkenes, such as perfluorocyclopentene and hexafluoropropene, were also reactive and could be transformed into hydrodefluorination products 5o:5o′ and 5p:5p′****, respectively, in reasonable yields (Figure 4).
Scope of group 13 fluoride catalyzed hydrodefluorination of fluorinated arenes and alkenes. aNMR yields measured by 19F NMR spectroscopy using 1,2-difluorobenzene as an internal standard. Isolated yields are in parentheses. bProduct formed alongside 12% 5l. cMe2PhSiH was used as a terminal reductant. dPh3SiH used as terminal reductant. e20 mol % catalyst loading used.
For comparison, contemporary approaches in main-group-catalyzed hydrodefluorination of fluoroarenes include catalysts based on P(III)/P(V) and Bi(I)/Bi(III) redox couples.^37−40^ The nucleophilic silicate complex TBAT has also been reported as a catalyst for hydrodefluorination of fluoroarenes with loadings as low as 0.1 mol % at 60 °C.^26^ Transition metal catalysts typically operate with a broader substrate range and improved efficiencies.^41^
The addition of 1-Al to NaBAr^F^4 in fluorobenzenes suggests an interaction between the two precatalyst components (Scheme 2). Hence, at 298 K, a 50 mM solution of a 1:1 mixture of 1-Al and NaBAr^F^4 displayed a defined and sharp ^19^F NMR resonance at δ = −180.2 ppm. 1-Al alone demonstrates a resonance at δ = −174.5 ppm at the same concentration. Varying the ratio of NaBAr^F^4:1-Al from 0.5:1 to 2:1 suggests that the binding event is 1:1 as the maximum Δδ ≈ 6 ppm is observed at this ratio with only small changes in chemical shift occurring at higher ratios. The reaction between 1-Al and NaBAr^F^4 was investigated at nine different temperatures across a 233–313 K temperature range, with consistent results supporting 1:1 binding at all temperatures (Figure 5a). No attempts were made to quantify these data, as it is likely that the equilibria at play are complicated by the potential for the solvent, fluorobenzene, to act as a competitive ligand for Na^+^. A similar reaction between a 1:1 mixture of 1-Ga and NaBAr^F^4 at 298 K in fluorobenzene displayed an upfield shift of the ^19^F NMR resonance Δδ ≈ 2.5 ppm relative to 1-Ga.
Proposed Reversible Reaction between 1-Al or 1-Ga and NaBArF4
(a) VT 19F NMR spectroscopic data from the reaction of 1-Al and NaBArF4 at different Al:Na ratios. (b) Calculated structure of 1-Ga-NaBArF4.
While the ^19^F NMR spectroscopic data support changes to the magnetic environment at fluorine upon the reaction of 1-Al or 1-Ga with NaBAr^F^4, these data in isolation do not give definitive information on the location of the Na^+^ ion within 1-Al-NaBAr^F^4 or 1-Ga-NaBAr^F^4. To probe the binding event, gallium hydride fluoride complex 6-Ga was prepared and reacted with NaBAr^F^4 in the hope that the ^2^JH–F coupling constant may shed light on changes to the Ga–F bonding on coordination to Na^+^. 6-Ga displays a doublet resonance at δ = −182.0 ppm (d, ^2^JH–F = 91.1 Hz) in the ^19^F NMR spectrum. The reaction with NaBAr^F^4 generates a new species assumed to be 6-Ga-NaBAr^F^4 with δ = −197.3 ppm (d, ^2^JH–F = 102.6 Hz). The latter demonstrates the expected upfield shift of the fluoride ligand Δδ ≈ 15 ppm, along with an increase of the ^2^JH–F coupling constant by more than 10 Hz, consistent with changes to bonding of the {GaHF} motif on association with Na^+^. Transition metal fluoride complexes are known to be effective hydrogen bond (and halogen bond) donors.^42−44^ Examples of coordination of metal fluorides to s-block cations are, however, less common. DFT calculations were undertaken to understand the nature of the binding event. Coordination of NaBAr^F^4 to 1-Al was calculated to be exergonic by ΔG°(298 K) = −1.4 kcal mol^–1^, consistent with a reversible binding event. A similar coordination geometry and an exergonic reaction ΔG°(298 K) = −2.9 kcal mol^–1^ was calculated for the reaction of NaBAr^F^4 with 1-Ga (Figure 5b). NBO and AIM calculations support the characterization event as a weak electrostatic interaction between the fluorine atoms and Na^+^ center (see the Supporting Information, Section 6.4).
A plausible reaction mechanism for the hydrodefluorination of fluoroarenes involves (i) σ-bond metathesis between Et_3_SiH and the group 13 fluoride 1-M forming the group 13 hydride complex and (ii) nucleophilic attack of the hydride on the fluorinated arene by a concerted S_N_Ar mechanism, liberating the hydrodefluorinated product and regenerating the group 13 fluoride (M= Al, Ga). We have previously shown that a β-diketiminate-stabilized aluminum dihydride complex is an effective reagent for the hydrodefluorination of electron-deficient arenes under forcing conditions.^23^
Further experiments and calculations were undertaken to shed light on the role of NaBAr^F^4 during catalytic hydrodefluorination. 1-Ga and 1-Ga-NaBAr^F^4 show different reactivities with Et_3_SiH. While no apparent reaction is observed between 1-Ga and Et_3_SiH up to 80 °C in fluorobenzene, the addition of 1 equiv of NaBAr^F^4 promotes the formation of 7-Ga and Et_3_SiF (δ = – 176.1 ppm) after 6 h at 80 °C (Figure 6a). The expected kinetic product of this reaction, 6-Ga, was shown to be in equilibrium with 1-Ga and 7-Ga at 25 °C in fluorobenzene (Figure 6b). DFT calculations support the role of NaBAr^F^4 in lowering the barrier for metathesis through fluoride coordination (Figure 6d). The reaction of 1-Ga-NaBAr^F^4 with Et_3_SiH was calculated to occur through TS-1 (ΔG^‡^298 K = 28.8 kcal mol^–1^), leading to the direct formation of 6-Ga-NaBAr^F^4. In the absence of the promoter, 1-Ga was calculated to react with Et_3_SiH by a higher barrier through TS-1’ (ΔG^‡^298 K = 30.5 kcal mol^–1^) to form 6-Ga. The lower energy of TS-1 relative to TS-1’ is explained through Na^+^ playing a role in charge stabilization during the polarization and breaking of the Ga---F bond.
(a) Stoichiometric reactions between 1-Ga and Et3SiH with and without NaBArF4. (b) Lithium-based exchange between 1-Ga, 7-Ga, and 6-Ga. (c) Hydrodefluorination of pentafluorobenzene catalyzed by 7-Ga and NaBArF4. (d) DFT calculated barriers for fluoride to hydride ligand metathesis for 1-Ga with and without NaBArF4. wB97xD/def2TZVPP/PCM(fluorobenzene) // wB97xD/6–31G*/6–311+G*/SDDAll/PCM(fluorobenzene). Gibbs energies are given in kcal mol–1.*
Experimentally, it was found that 7-Ga is an effective nucleophile for the hydrodefuorination of pentafluoropyridine at 100 °C and that 10 mol % 7-Ga+ NaBAr^F^4 can be used as a precatalyst for the hydrodefluorination of the same substrate with Et_3_SiH at 160 °C (Figure 6c).^45^ These findings suggest a synergistic role for Na^+^ and the group 13 complex in catalysis and are reminiscent of cooperative effects in not only the stoichiometric activation of fluoroarenes with heterometallic complexes but also catalytic applications of s/p heterobimetallic complexes in hydrogenation, hydroamination, hydrophosphination, and hydroboration of unsaturated substrates.^46,47^ Based on the current data, however, we cannot exclude alternative roles for NaBAr^F^4 in catalysis, including modification of the solvent polarity, or in the S_N_Ar step, due to coordination to the substrate and polarization of the reacting carbon–fluorine bond.
Conclusions and Summary
In summary, a series of four-coordinate group 13 fluoride complexes (M = B, Al, Ga) are reported as catalysts for the thiodefluorination and hydrodefluorination of poly- and perfluoroarenes using silanes as terminal reagents. The scope of reactivity in the electrophile is broad, provided that the aromatic ring is electron-deficient. For thiodefluorination, mechanistic studies support a plausible role for group 13 thiolate intermediates, which are capable of C–S bond formation through nucleophilic attack on the aromatic ring. Similarly, for hydrodefluorination, group 13 hydrides are likely key catalytic intermediates. In both cases, catalytic turnover requires metathesis (ligand exchange) between the group 13 complex and silicon reagent; a step that requires breaking of strong group 13 metal–fluoride bonds. In the case of thiodefluorination, RS^–^-to-F^–^ metathesis occurs in the absence of an additive, while in the case of hydrodefluorination, H^–^-to-F^–^ exchange requires the use of NaBAr^F^4 as an additive. Stoichiometric experiments and DFT calculations are consistent with NaBAr^F^4 playing a role in activating and weakening the group 13 metal–fluoride bond through M–F---Na^+^ interactions, facilitating metathesis with silane reagents. This additive effect may have important implications for future catalyst design.^48^
Experimental Section
General Procedure for Thiodefluorination of Arenes
In a glovebox, the fluoroarene (0.05 mmol), Me_3_Si-X (Me_3_Si = trimethylsilane, X = SPh) reagent (0.15–0.5 mmol), catalyst (10–20 mol % 1-B, 1-Al, or 1-Ga), and PhCF_3_ or PhF (dry and degassed) were added to a J. Young’s NMR tube and sealed. The total volume of the PhCF_3_ and C_6_D_6_ (5:1) or PhF and C_6_D_6_ (5:1) solution was made up of 0.6 mL. The reaction was monitored by ^19^F NMR spectroscopy and, depending on the substrate, heated between 60 and 160 °C (Figure 2). 1,2-Difluorobenzene in a sealed glass capillary containing C_6_D_6_ (δ = 138.3 ppm) was added as an internal standard, and the reaction mixture was analyzed by quantitative ^19^F NMR spectroscopy. Nonvolatile products were isolated and purified by column chromatography. For full details and characterization data on 2a–m, see the Supporting Information.
General Procedure for Hydrodefluorination of Fluoroarenes
In a glovebox, the fluoroarene (0.05 mmol), silane reagent (0.15–0.5 mmol), catalyst mixture (10–20 mol % 1-B, 1-Al, or 1-Ga + 10–20 mol % NaBAr^F^4), and PhCF_3_ or PhF (dry and degassed) were added to a J-Young’s NMR tube and sealed. The total volume of the PhCF_3_ and C_6_D_6_ (5:1) or PhF and C_6_D_6_ (5:1) solution was made up to 0.6 mL. The reaction was monitored by ^19^F NMR spectroscopy and, depending on the substrate, heated between 80 and 160 °C (Figure 4). 1,2-Difluorobenzene in a sealed glass capillary containing C_6_D_6_ (δ = 138.3 ppm) was added as an internal standard, and the reaction mixture was analyzed by quantitative ^19^F NMR spectroscopy. Nonvolatile products were isolated and purified by column chromatography. For full details and characterization data on 5a–p, see the Supporting Information.
Computational Methods
DFT calculations were performed using Gaussian 09 (Revision D.01) using an ultrafine integration grid (int = ultrafine). Geometry optimizations and frequency calculations were performed using the ωB97xD density functional, including solvent corrections (PCM, fluorobenzene) with SDDAll (Na, Ga), 6-31G** (C, H) and 6-311+G* (N, F, Si) basis sets. Frequency analyses for all stationary points were performed using the enhanced criteria to confirm the nature of the structures as either minima (no imaginary frequency) or transition states (only one imaginary frequency). The electronic energies of the optimized geometries were calculated using the ωB97xD functional with solvent corrections (PCM, fluorobenzene) using the def2TZVPP basis sets for all atoms. The Gibbs free energy correction from the frequency calculation was added to this electronic energy to generate Gibbs free energy values (calculated at 298.15 K, 1 atm) for the stationary points. Intrinsic reaction coordinate calculations were used to connect transition states and minima located on the potential energy surface. NBO analysis was performed at the ωB97xD/def2TZVPP level using NBO 6.0 and is stated as such. QTAIM analysis was conducted by using the AIMAll package.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Erdmann P.; Leitner J.; Schwarz J.; Greb L. An Extensive Set of Accurate Fluoride Ion Affinities for p-Block Element Lewis Acids and Basic Design Principles for Strong Fluoride Ion Acceptors. Chem Phys Chem 2020, 21, 987–994. 10.1002/cphc.202000244.32212357 PMC 7317340 · doi ↗ · pubmed ↗
- 2Olah G. A.; Kuhn S. J. Selective Friedel—Crafts Reactions. I. Boron Halide Catalyzed Haloalkylation of Benzene and Alkylbenzenes with Fluorohaloalkanes. J. Org. Chem. 1964, 29, 2317–2320. 10.1021/jo 01031 a 051. · doi ↗
- 3Caputo B. C.; Stephan D. W. Activation of Alkyl C–F Bonds by B(C 6F 5)3: Stoichiometric and Catalytic Transformations. Organometallics 2012, 31, 27–30. 10.1021/om 200885 c. · doi ↗
- 4Petrov V. A.; Krespan C. G.; Smart B. E. Electrophilic Reactions of Fluorocarbons under the Action of Aluminum Chlorofluoride, a Potent Lewis Acid. J. Fluorine Chem. 1996, 77, 139–142. 10.1016/0022-1139(96)03391-X. · doi ↗
- 5Ahrens M.; Scholz G.; Braun T.; Kemnitz E. Catalytic Hydrodefluorination of Fluoromethanes at Room Temperature by Silylium-ion-like Surface Species. Angew. Chem., Int. Ed. 2013, 52, 5328–5332. 10.1002/anie.201300608.23589445 · doi ↗ · pubmed ↗
- 6Meißner G.; Dirican D.; Jäger C.; Braun T.; Kemnitz E. Et 3Ge H versus Et 3Si H: Controlling Reaction Pathways in Catalytic C–F Bond Activations at a Nanoscopic Aluminum Chlorofluoride. Catal. Sci. Technol. 2017, 7, 3348–3354. 10.1039/C 7CY 00845 G. · doi ↗
- 7Meißner G.; Kretschmar K.; Braun T.; Kemnitz E. Consecutive Transformations of Tetrafluoropropenes: Hydrogermylation and Catalytic C–F Activation Steps at a Lewis Acidic Aluminum Fluoride. Angew. Chem., Int. Ed. 2017, 56, 16338–16341. 10.1002/anie.201707759.28981989 · doi ↗ · pubmed ↗
- 8Pan X.; Talavera M.; Scholz G.; Braun T. Chlorodefluorination of Fluoromethanes and Fluoroolefins at a Lewis Acidic Aluminum Fluoride. Chem Cat Chem. 2022, 14, e 20220002910.1002/cctc.202200029. · doi ↗