MYD88‐mutated Lymphoplasmacytic Lymphoma With Monoclonal Immunoglobulin G: A Case Report
Morten Yung Isaksen, Olav Karsten Vintermyr, Håkon Reikvam

TL;DR
A rare case of lymphoplasmacytic lymphoma secreting IgG instead of IgM is reported, highlighting the importance of considering this diagnosis even without typical IgM markers.
Contribution
This case report expands the understanding of lymphoplasmacytic lymphoma by presenting an atypical IgG-secreting variant.
Findings
The patient had LPL secreting monoclonal IgG instead of the usual IgM.
The patient was treated with bortezomib, dexamethasone, and rituximab due to disease progression.
This case emphasizes the need to consider LPL in differential diagnosis even without IgM.
Abstract
Lymphoplasmacytic lymphomas (LPL) are usually associated with serum monoclonal immunoglobulin M (IgM). Nevertheless, in some cases, these cells may secrete IgA or IgG monoclonal proteins or remain non‐secretory. We report a case from a patient with LPL‐secreting IgG who developed anaemia and splenomegaly during the disease course that necessitated treatment with bortezomib, dexamethasone, and rituximab. The case illustrates the need for clinicians and pathologists to consider LPLs as a differential diagnosis also without a serum monoclonal IgM. Clinical Trial Registration: The authors have confirmed clinical trial registration is not needed for this submission.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
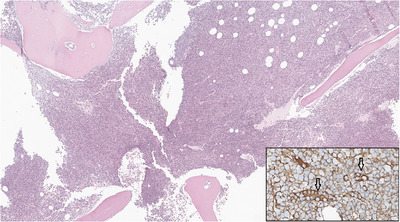 FIGURE 1
FIGURE 1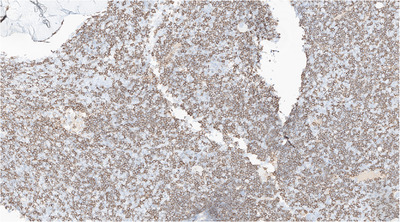 FIGURE 2
FIGURE 2| Value | Reference | Diagnosis | Initiating treatment | Ending treatment |
|---|---|---|---|---|
| Haemoglobin | 11.7–15.3 g/dL | 12.8 | 9.0 | 11.4 |
| WBC | 4.1–9.8 × 109/L | 7.4 | 19.8 | 5.6 |
| Platelets | 165–387 × 109/L | 202 | 79 | 172 |
| Creatinine | 45–90 µg/L | 65 | 97 | 86 |
| LDH | 115–255 U/L | 191 | 189 | 183 |
| IgG | 6.00–15.3 g/L | 45.0 | 39.7 | 28.2 |
| IgA | 0.80–4.00 g/L | 0.5 | 0.35 | 0.84 |
| IgM | 0.30–2.30 g/L | 0.2 | 0.57 | 1.27 |
| M‐protein | 0.0 g/L | — | 21.7 | 7.5 |
| Kappa light chains | 8.30–27.0 mg/L | — | 649 | 382 |
| Lambda light chains | 0.31–1.56 mg/L | — | 72.9 | 67.5 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsChronic Lymphocytic Leukemia Research · Lymphoma Diagnosis and Treatment · Viral-associated cancers and disorders
Introduction
1
Lymphoplasmacytic lymphomas (LPLs) are indolent B‐cell lymphomas. Most LPLs are associated with clonal lymphoplasmacytic cells secreting immunoglobulin M (IgM), referred to as IgM‐LPL or Waldenström macroglobulinaemia (WM) type. Only about 5% of LPLs are non‐WM type, and cases with monoclonal IgG make up a subset of these. Other non‐WM type LPLs are IgA‐LPLs, non‐secretory LPLs and IgM LPLs with only extramedullary involvement [1].
Compared to WM, non‐WM type LPLs have been shown to present with extramedullary disease more often than WM, while they harbour the MYD88‐mutation less frequently than their IgM‐secreting counterpart [2, 3, 4]. Asymptomatic cases are followed with regular observation, while patients with findings suggestive of significant infiltration of the bone marrow or other organs, like cytopenias, splenomegaly, or bulky lymphadenopathy, should be offered therapy. Non‐WM type LPLs are generally treated similarly to classical WM types, with chemoimmunotherapy or a Bruton tyrosine kinase inhibitor [3, 4, 5].
Case Presentation
1.1
A woman in her 60s was referred to the haematology department due to an elevated erythrocyte sedimentation rate and the presence of a serum IgG monoclonal protein, type kappa. Blood values upon referral are given in Table 1. Bone marrow smear microscopy, performed under suspicion of multiple myeloma, revealed no plasma cells, although it showed a marked increase in lymphocytes, constituting 67% of nucleated cells. Given the findings, lymphoma was suspected. Multiparametric flow cytometry of bone marrow aspirate confirmed a clonal expansion of a distinct cell population with the following immune phenotype: CD45+/19+/20++/23‐/22dim/5‐/10+/38‐/11c‐/kappa light chain positive, comprising approximately 33% of nucleated cells. A subsequent trephine biopsy demonstrated widespread marrow infiltration by lymphoplasmacytic cells in a diffuse and interstitial pattern (Figure 1‐2). Based on these findings, a diagnosis of lymphoplasmacytic lymphoma with monoclonal IgG was established. Further genetic testing revealed the presence of the MYD88 L265P mutation, supporting the diagnosis.
Trephine biopsy showing a hypercellular bone marrow (HE x3). Inset: Plasma cells staining positive for IgG (arrows; IHC x40). HE, haematoxylin‐eosin; IHC, immunohistochemistry.
Trephine biopsy with a marked increase of CD20‐positive cells consistent with B‐cells (IHC x8). IHC, immunohistochemistry.
The patient did not have any cytopenias, lymphadenopathy or constitutional symptoms at the time of diagnosis. She was followed untreated with regular visits at the outpatient clinic over several years. More than 5 years after initial diagnosis her haemoglobin levels started to drop, with a slowly worsening anaemia developing over a period of another 7 years. The blood samples at this time are given in Table 1. In addition, the patient had a computed tomography scan of the neck, thoracic‐, abdominal‐, and pelvic cavity, which showed an enlarged spleen with a diameter of 21 cm at the most and a couple of slightly enlarged lymph nodes in the mediastinum. A repeated trephine biopsy was done with similar findings as at the time of initial diagnosis.
The patient had symptoms of anaemia and discomfort from her enlarged spleen, and hence indication for starting therapy for her LPL was found. Treatment with bortezomib, dexamethasone, and rituximab (BDR) as first‐line treatment was initiated. After six cycles of BDR the patient was in good clinical condition. Her peripheral haematological values had almost normalized (Table 1). M protein levels were reduced to 7.5 g/L consistent with a partial response to therapy. The patient is currently followed untreated with regular visits at the outpatient clinic. The M protein levels are gradually rising, although she has so far not developed any signs indicating the need for second‐line therapy.
Discussion
2
LPLs are rare lymphomas, making up approximately 2% of haematological malignancies in Europe and the United States annually [6, 7]. As a rare subgroup, LPLs associated with monoclonal proteins other than IgM make up only a small fraction of the total cases of LPLs. Previous case series and register‐based studies have shown clinical differences between non‐WM LPL and WM, but the total number of patients with non‐WM LPL is small, and there is considerable heterogeneity between findings in the reports. When comparing cases of non‐WM LPL to WM, several studies found significantly more extramedullary symptoms in patients with non‐WM LPL, while the MYD88‐mutation was less frequently found in these patients. On the other hand, there were inconsistent findings regarding differences in age, sex and laboratory findings at the time of diagnosis across the non‐WM LPL and WM groups [2–4, 9, 10]. Due to the natural absence of elevated levels of the high molecular weight IgM‐pentamer, patients with non‐WM LPL rarely show symptoms of hyperviscosity.
In our patient, the findings of widespread marrow infiltration by lymphoplasmacytic cells in the trephine biopsy suggested an LPL. Alternative diagnoses were considered, like marginal zone lymphoma and follicular lymphoma, but immunohistochemical findings were not consistent with either of these. The finding of a MYD88 L265P mutation in our patient also supported the diagnosis of an LPL. The patient was not tested for CXCR4‐mutations, the second most common somatic mutation in patients with WM.
Our patient developed symptomatic splenomegaly and anaemia more than 10 years after the initial diagnosis. Due to its rarity, there are no guidelines specific to the treatment of non‐WM LPLs, and they are often treated similarly to WM. Several reports comparing non‐WM type LPLs to WM have found a shorter time from diagnosis to first treatment for patients with non‐WM type LPLs [2, 3].
For our patient, a regimen consisting of BDR was chosen above bendamustine and rituximab, as the indication for therapy was bone marrow failure, and the BDR regime is regarded as less myelosuppressive [8]. She showed good tolerance for treatment with BDR and is currently feeling well more than 18 months after finishing therapy.
Overall survival (OS) in patients with non‐WM type LPLs seems to be similar to OS in patients with WM [2, 3, 9]. Even though non‐WM LPLs share several features with classical WM, this subtype of LPL is not well understood. Further research is warranted to discriminate unique clinical, pathological and molecular features of non‐WM type LPL, as well as disease‐specific therapy and follow‐up.
Author Contributions
All authors have contributed to the manuscript and approved the final version.
Conflicts of Interest
The authors have nothing to report.
Patient Consent Statement
Supplied upon request
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1R. Alaggio , C. Amador , I. Anagnostopoulos , et al., “The 5th Edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms,” Leukemia 36, no. 7 (2022): 1720–1748.35732829 10.1038/s 41375-022-01620-2PMC 9214472 · doi ↗ · pubmed ↗
- 2M. Varettoni , E. Boveri , S. Zibellini , et al., “Clinical and Molecular Characteristics of Lymphoplasmacytic Lymphoma Not Associated With an Ig M Monoclonal Protein: A Multicentric Study of the Rete Ematologica Lombarda (REL) Network,” American Journal of Hematology 94, no. 11 (2019): 1193–1199.31378966 10.1002/ajh.25600 · doi ↗ · pubmed ↗
- 3J. J. Castillo , G. Itchaki , J. N. Gustine , et al., “A Matched Case‐control Study Comparing Features, Treatment and Outcomes Between Patients With Non‐Ig M Lymphoplasmacytic Lymphoma and Waldenström Macroglobulinemia,” Leukemia & Lymphoma 61, no. 6 (2020): 1388–1394.31992103 10.1080/10428194.2020.1719100 · doi ↗ · pubmed ↗
- 4X. Cao , L. J. Medeiros , Y. Xia , et al., “Clinicopathologic Features and Outcomes of Lymphoplasmacytic Lymphoma Patients With Monoclonal Ig G or Ig A Paraprotein Expression,” Leukemia & Lymphoma 57, no. 5 (2016): 1104–1113.26421453 10.3109/10428194.2015.1096357 · doi ↗ · pubmed ↗
- 5M. A. Dimopoulos and E. Kastritis , “How I Treat Waldenström Macroglobulinemia,” Blood 134, no. 23 (2019): 2022–2035.31527073 10.1182/blood.2019000725 · doi ↗ · pubmed ↗
- 6L. M. Morton , S. S. Wang , S. S. Devesa , P. Hartge , D. D. Weisenburger , and M. S. Linet , “Lymphoma Incidence Patterns by WHO Subtype in the United States, 1992–2001,” Blood 107, no. 1 (2006): 265–276.16150940 10.1182/blood-2005-06-2508 PMC 1895348 · doi ↗ · pubmed ↗
- 7M. Sant , C. Allemani , C. Tereanu , et al., “Incidence of Hematologic Malignancies in Europe by Morphologic Subtype: Results of the HAEMACARE Project,” Blood 116, no. 19 (2010): 3724–3734.20664057 10.1182/blood-2010-05-282632 · doi ↗ · pubmed ↗
- 8S. P. Treon , L. Ioakimidis , J. D. Soumerai , et al., “Primary Therapy of Waldenström Macroglobulinemia With Bortezomib, Dexamethasone, and Rituximab: WMCTG Clinical Trial 05–180,” Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology 27, no. 23 (2009): 3830–3835.19506160 10.1200/JCO.2008.20.4677 PMC 2727288 · doi ↗ · pubmed ↗