Chiari malformation type 1 with combined nuclear PAX1 and DKK1 genes and mitochondrial D-loop variants: a case report
Siti Nornazihah Mohd Rosdi, Suzuanhafizan Omar, Mazira Mohamad Ghazali, Ab Rahman Izaini Ghani, Abdul Aziz Mohamed Yusoff

TL;DR
This case report describes a 15-year-old girl with Chiari malformation type 1 and a unique combination of nuclear and mitochondrial genetic variants.
Contribution
The study reports the first case of CM 1 with combined nuclear PAX1/DKK1 and mitochondrial D-loop variants.
Findings
A silent mutation in PAX1 and a DKK1 intronic variant were identified in nuclear DNA.
Mitochondrial D-loop variants, including some not previously documented, were found.
The findings suggest a potential role of mitochondrial D-loop mutations in CM 1.
Abstract
Chiari malformation type 1 (CM 1) is a rare and complex neurological condition. This congenital condition affects the lower posterior fossa, where the brain connects to the spinal cord. Although the exact cause of CM 1 remains unclear, genetic predisposition plays a considerable role in structural defects of the cerebellum. Here, we report on a 15-year-old female patient with CM 1 who exhibited both nuclear and mitochondrial genetic variants, a combination that has not been previously described. We identified a silent mutation in exon 2 (c. 556 G>A, p. Lys185 = ) of PAX1 and a DKK1 variant in intron 3 (548-3 t > C) in the nuclear DNA. We also screened the D-loop region of mitochondrial DNA as it exhibits a higher susceptibility to mutations than other mitochondrial DNA regions. Several hotspot variants were revealed, including those in positions 303-309 and 16519 (t > C), as well as…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
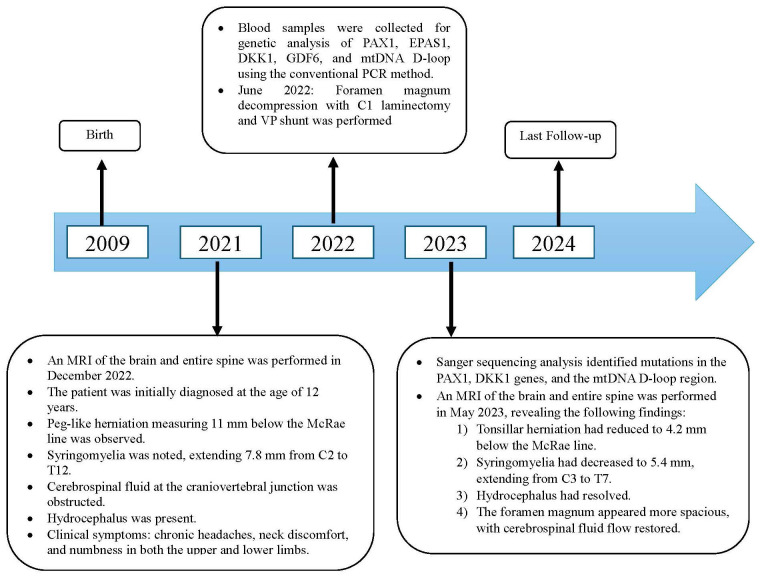 Figure 1
Figure 1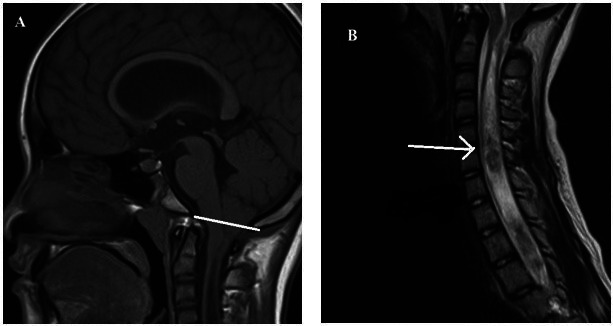 Figure 2
Figure 2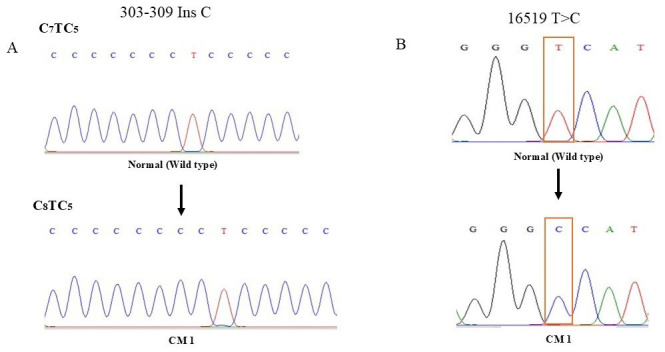 Figure 3
Figure 3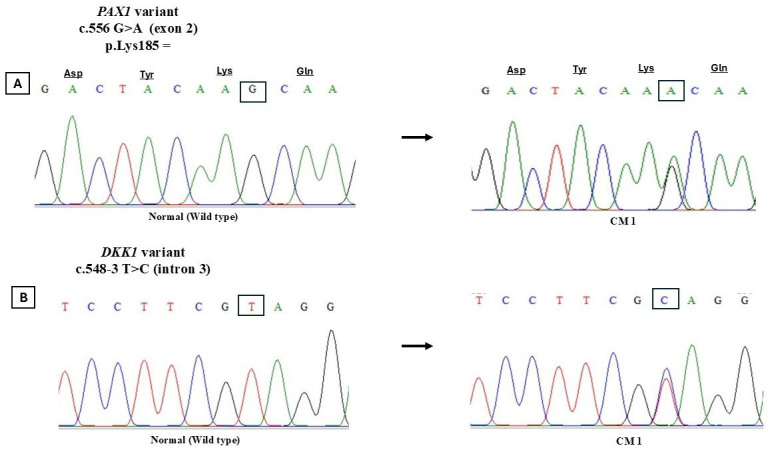 Figure 4
Figure 4| Somatic mutation | Homoplasmy/ heteroplasmy | Region | Novel/ reported | References |
|---|---|---|---|---|
| 210 A>G | homoplasmy | HVS2 | not reported in MITOMAP | present study |
| 303-309 ins C | homoplasmy | CSB2 | prostate cancer | ( |
| 489 | homoplasmy | HVS3 | prostate cancer | ( |
| 16031 C>G | homoplasmy | HVS1 | not reported in MITOMAP | present study |
| 16145 G>A | homoplasmy | HVS1 | not reported in MITOMAP | present study |
| 16181 A>G | homoplasmy | HVS1 | not reported in MITOMAP | present study |
| 16192 C>T | homoplasmy | HVS1 | colonic mucosa | ( |
| 16291 C>T | homoplasmy | HVS1 | diabetes type 2 | ( |
| 16304 | homoplasmy | HVS1 | prostate cancer | ( |
| 16519 | homoplasmy | ATT | prostate cancer | ( |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSpinal Dysraphism and Malformations
Chiari malformation type 1 (CM 1) is a neurological condition characterized by the herniation of the cerebellar tonsils more than 5 mm through the foramen magnum (1). It is often associated with syringomyelia and is more prevalent in women (2). CM 1 can be asymptomatic or present with neurological symptoms, including headaches (98% of patients), ocular disturbances, neck pain, vertigo, numbness, and scoliosis. Surgical treatment is required if severe neurological symptoms are present.
The cause of CM 1 remains unclear, but familial clustering and hereditary influence support the role of genetic factors (3). However, no single gene mutation has been identified as the primary cause of cerebellar defects. Environmental factors may also contribute.
Mitochondria house their own genetic material, mitochondrial DNA (mtDNA), which begins to replicate at the displacement-loop (D-loop), a non-coding region where two DNA strands are separated and stabilized by a third displaced strand (4). This D-shaped structure, evident in electron micrographs, also plays a crucial role in repairing DNA double-strand breaks through homologous recombination (4). The D-loop is a significant hotspot for genetic alterations in human cancers, which may impair mtDNA replication or transcription, resulting in mitochondrial dysfunction and increased cellular production of reactive oxygen species (ROS) (5).
The role of mtDNA in CM 1 remains incompletely understood and has not been previously explored in genetic pathways related to the condition. Given the rarity and complexity of CM 1, we focused on the D-loop region of mtDNA due to its higher mutation susceptibility. This additional testing aimed to provide a detailed genetic analysis, potentially shedding new insights on the genetic causes of CM 1 and guiding future research. Therefore, we report on a CM 1 patient with combined nuclear DNA (PAX1 and DKK1 genes) and mtDNA variants, which has not been previously documented in literature or reference laboratory databases.
CASE REPORT
A 15-year-old female patient (Figure 1), first diagnosed in 2021 at the age of 12, was evaluated at the Neuroscience Specialist Clinic of Universiti Sains Malaysia Specialist Hospital due to a history of chronic headache, neck discomfort, and bilateral upper and lower limb numbness. Magnetic resonance imaging (MRI) of the brain and entire spine revealed a peg-like herniation of the cerebellar tonsils through the foramen magnum, measuring 11 mm. The herniation led to crowding of the foramen magnum, with associated hydrocephalus and syringomyelia. The MRI findings confirmed the diagnosis of CM 1 (Figure 2). The MRI of the cervical spine revealed a long segment of hypointensity on T1/flair at the center of the spinal cord, which extended from the C2 level downward. It was consistent with cerebrospinal fluid (CSF) signal intensity and indicated a syrinx. Subsequently, the patient underwent a right ventriculoperitoneal shunt and foramen magnum decompression with C1 laminectomy. Postoperative MRI revealed a regression of tonsillar herniation to 4.2 mm below the McRae line, along with a reduction in syringomyelia to 5.4 mm. Additionally, hydrocephalus had resolved. The patient remains under regular follow-up.
The timeline of events and interventions. mtDNA – mitochondrial DNA; PCR – polymerase chain reaction; VP – ventriculoperitoneal; MRI – magnetic resonance imaging.
(A) Sagittal magnetic resonance imaging (MRI) showing Chiari malformation 1 (CM 1). The white line represents the McRae line connecting the basion and opisthion. (B) T2-weighted sagittal MRI displaying a cervical-thoracic syrinx, indicated by the white arrow.
Detection of mtDNA D-loop variants
After providing informed consent, the patient underwent mtDNA D-loop mutation screening. Peripheral blood was collected for genomic DNA extraction, followed by the amplification of the mtDNA D-loop region using polymerase chain reaction (PCR) with human mitochondrial primers (5). Sanger sequencing was performed with the Big Dye Terminator cycle sequencing kit (Applied Biosystems, Foster City, CA, USA), and the data were analyzed with an Applied Biosystems 3730 Series Genetic Analyzer. Mutations and polymorphisms were confirmed through a repeated analysis of both strands. The results were compared with the Cambridge Reference Sequence of human mtDNA (NC_012920) from the MITOMAP database (http://www.mitomap.org). The screening revealed multiple mtDNA D-loop variants, such as those at positions 303-309 and 16519 (t > C), along with some variants not reported in MITOMAP (Table 1). Typical electropherograms are shown in Figure 3.
Mitochondrial DNA D-loop mutations in a patient with Chiari malformation 1. (A) The insertion of a cytosine at nucleotide position 303-309. (B) A homoplasmic mutation involving a T-to-C transition at nucleotide position 16519.
Detection of PAX1 and DKK1 gene variants
Genomic DNA was extracted with the same method for variant analysis in PAX1 and DKK1 genes, with PCR primers and conditions obtained from McGaughran et al (9) and Marello et al (3), respectively. Sanger sequencing identified a synonymous transition mutation (c.556 G>A) in the PAX1 gene at codon 185 (AAG) in exon 2, maintaining the amino acid lysine (Figure 4). A heterozygous variant (c.548-3 t > C) was detected in the intron of the DKK1 gene.
(A) Heterozygous PAX1 variant in exon 2, c.556 G>A (p. Lys185 = ). (B) DKK1 variant in intron 3, 548-3 t > C.
DISCUSSION
CM 1 is largely sporadic, though familial aggregation and hereditary influence suggest a genetic predisposition. A contributing factor may be the underdevelopment of occipital somites from the paraxial mesoderm, involving the PAX gene family (10,11). PAX genes encode transcription factors critical for pattern formation during vertebrate embryogenesis. Specifically, PAX1, located on chromosome 20p11.2, is essential for vertebral segmentation and development (9). A disrupted PAX1 function has been associated with vertebral fusion and re-segmentation issues (9). In this study, we identified a silent mutation in exon 2 of PAX1 (c. 556 G>A), which did not alter protein structure. However, silent mutations may still impact protein levels and function by influencing mRNA stability, splicing, or translation efficiency (12). Macaya et al reported a de novo silent mutation (c.3612 TCA>TCC [p. Ser1204Ser]) in Treacher Collins syndrome, an autosomal dominant craniofacial disorder linked to the TCOF1 gene. This mutation caused splicing defects and abnormal mRNA processing (13).
DKK1, a 29 kDa protein in the Dickkopf (DKK) family, inhibits the Wnt signaling pathway, which is critical for vertebrate embryogenesis, including head formation, skeletal development, and limb patterning (14). It encodes a secreted ligand that binds to LRP6 receptors, blocking Wnt signaling and pathway activation (15). Disruptions in this pathway can lead to craniofacial and neural malformations that contribute to CM 1. An altered DKK1 expression affected craniofacial and central nervous system (CNS) development in animal studies. For example, DKK1 overexpression in Xenopus embryos caused enlarged anterior head structures, while anti-DKK1 antibody injections led to microcephaly (16). Although the DKK1 intronic variant (Intron 3 c.548-43 t > C) identified in this study lacked functional evidence, its potential impact on other cell types or developmental stages cannot be ruled out. Intronic mutations can become pathogenic if they activate cryptic splice sites, leading to exon skipping or intronic sequence inclusion in mature transcripts (9). An example includes β-thalassemia, where an intronic mutation disrupts pre-mRNA splicing, impairing β-globin protein production (17).
Currently, no studies have examined mitochondrial genome mutations associated with CM 1. Research on CM 1 has primarily focused on nuclear DNA due to its established role in genetic predispositions influencing CM 1 development. This study also concentrated on mtDNA, often referred to as the second genome, targeting the D-loop region for its high mutation susceptibility (5). In addition to PAX1 and DKK1 variants, the patient exhibited mtDNA D-loop mutations at positions 309 and 161519. The D-loop, a non-coding regulatory region spanning 1124 bp (position 16024-576), controls mtDNA replication and transcription, containing hypervariable regions HVI (16024-16383), HVII (57-372), and HVIII (438-574) (5). This region is a hotspot for mutations linked to various disorders, including cancers and neurodegenerative diseases. Notably, the C7TC5 mononucleotide repeat sequence (303-316/318) frequently exhibits deletion or insertion mutations, termed mitochondrial microsatellite instability. mtDNA mutates over ten times faster than nuclear DNA due to its proximity to the electron transport chain and the absence of protective histone proteins, which increases its vulnerability to ROS (5). These mutations impair ATP synthesis and elevate ROS production, inducing oxidative stress and mitochondrial dysfunction. Understanding mtDNA's involvement in CM 1 may allow insights into its pathogenic mechanisms.
In the context of CM 1, mitochondrial dysfunction can disrupt the development or stability of brain structures, including the cerebellum. Mutations in the mtDNA D-loop may increase cellular ROS production, contributing to oxidative stress in neural cells. This could, in turn, affect mtDNA replication and transcription.
In conclusion, we present the first reported case of a patient with CM 1 exhibiting a dual genetic variant (nDNA and mtDNA), which has not been previously documented in literature or reference laboratory databases. This case highlights a potential for further investigation into the relationship between mtDNA and CM 1, suggesting a new direction for further research. CM 1 is closely linked to CNS anomalies, as it involves the structural displacement of cerebellar tonsils into the spinal canal. Furthermore, mitochondrial dysfunction associated with alterations in the mtDNA D-loop region may play a role in CNS anomalies, potentially disrupting energy metabolism and increasing oxidative stress levels.
This case report enhances our understanding of CM 1 by exploring the involved genetic factors, particularly nuclear and mitochondrial variants, and highlighting the role of mtDNA mutations in the development of the condition. These findings could guide future research into genetic predisposition and potential therapeutic strategies.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Mueller DM Oro’JJ Prospective analysis of presenting symptoms among 265 patients with radiographic evidence of Chiari malformation type I with or without syringomyelia. J Am Acad Nurse Pract 2004 16 134 8 10.1111/j.1745-7599.2004.tb 00384.x 15130068 · doi ↗ · pubmed ↗
- 2Zhang Y Zhang N Qiu H Zhou J Li P Ren M An efficacy analysis of posterior fossa decompression techniques in the treatment of Chiari malformation with associated syringomyelia. J Clin Neurosci 2011 18 1346 9 10.1016/j.jocn.2011.01.026 21782445 · doi ↗ · pubmed ↗
- 3Merello E Tattini L Magi A Accogli A Piatelli G Pavanello M Exome sequencing of two Italian pedigrees with non-isolated Chiari malformation type i reveals candidate genes for cranio-facial development. Eur J Hum Genet 2017 25 952 9 10.1038/ejhg.2017.71 28513615 PMC 5567145 · doi ↗ · pubmed ↗
- 4Szmyd B Stanisławska P Podstawka M Zaczkowski K Izbiński PM Kulczycka-Wojdala D D-loop mutations as prognostic markers in glioblastoma—a pilot study. Int J Mol Sci 2024 ••• 25 10.3390/ijms 25084334 38673919 PMC 11050196 · doi ↗ · pubmed ↗
- 5Mohamed Yusoff AA Nasir KNM Haris K Khair SZNM Ghani ARIA Idris Z Detection of somatic mutations in the mitochondrial DNA control region D-loop in brain tumors: The first report in Malaysian patients. Oncol Lett 2017 14 5179 88 10.3892/ol.2017.6851 29098023 PMC 5652220 · doi ↗ · pubmed ↗
- 6Chen JZ Gokden N Greene GF Mukunyadzi P Kadlubar FF Extensive somatic mitochondrial mutations in primary prostate cancer using laser capture microdissection. Cancer Res 2002 62 6470 4 12438238 · pubmed ↗
- 7Greaves LC Beadle NE Taylor GA Commane D Mathers JC Khrapko K Quantification of mitochondrial DNA mutation load. Aging Cell 2009 8 566 72 10.1111/j.1474-9726.2009.00505.x 19624578 PMC 2752485 · doi ↗ · pubmed ↗
- 8Saha SK Al Saba A Hasib M Al Rimon R Hasan I Alam MS Evaluation of D-loop hypervariable region I variations, haplogroups and copy number of mitochondrial DNA in Bangladeshi population with type 2 diabetes. Heliyon 2021 7 e 07573 10.1016/j.heliyon.2021.e 07573 34377852 PMC 8327661 · doi ↗ · pubmed ↗