Complete mitochondrial genomes of Sinonovacularivularis and Novaculinachinensis and their phylogenetic relationships within family Pharidae
Yiping Meng, Liyuan Lv, Zhihua Lin, Demin Zhang, Yinghui Dong

TL;DR
This paper analyzes the mitochondrial genomes of two bivalve species to clarify their evolutionary relationships and adaptations within the family Pharidae.
Contribution
The study provides new mitochondrial genome data and insights into the phylogeny and salinity adaptation of Pharidae bivalves.
Findings
N. chinensis has the smallest genome size but highest AT content among Pharidae mitogenomes.
Phylogenetic analysis confirms the monophyly of Solenoidea and close relationships within Pharidae.
Positive selection and divergent evolution were detected in nad5, suggesting adaptation to salinity.
Abstract
Pharidae is one of the most ecologically and commercially significant families of marine Bivalvia; however, the taxonomy and phylogeny of Pharidae has been ongoing for quite some time and remains a contentious issue. Here, to resolve some problematical relationships among this family, the complete mitochondrial genomes (mitogenomes) of Sinonovacularivularis (17,159 bp) and Novaculinachinensis (15,957 bp) were assembled, and a comparative mitochondrial genomic analysis was conducted. Both mitogenomes contain 12 protein-coding genes, 22 transfer RNA genes, and two ribosomal RNA genes. Among the published Pharidae mitogenomes, N.chinensis exhibited the smallest genome size but the highest AT content. The results of the phylogenetic trees confirmed the monophyly of the family Solenoidea, and indicated that N.chinensis and Sinonovacula (S.constricta and S.rivularis) were closely related in…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
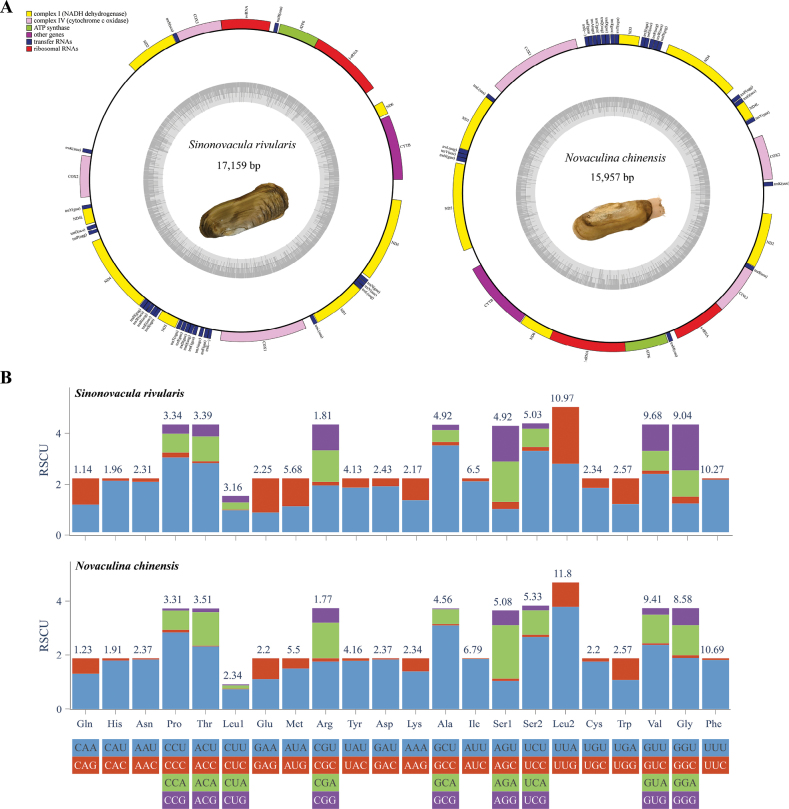 Figure 1
Figure 1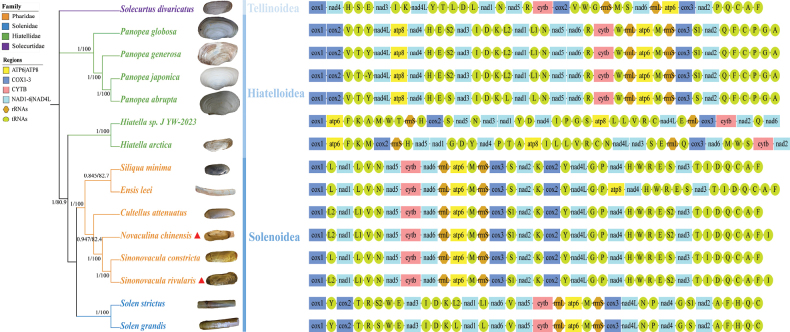 Figure 2
Figure 2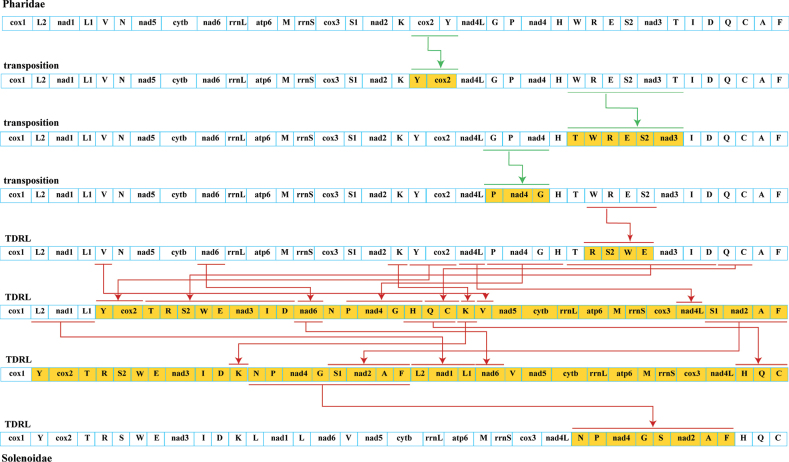 Figure 3
Figure 3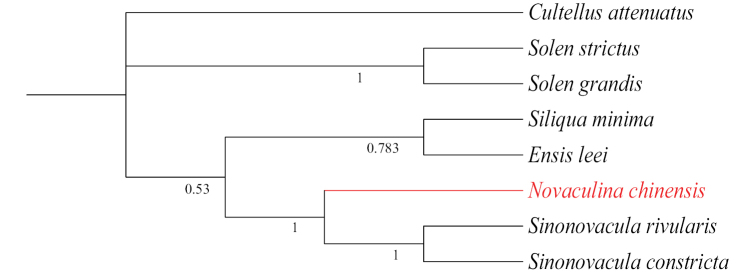 Figure 4
Figure 4 Figure 5
Figure 5| Order | Superfamily | Family | Species | Length (bp) | Accession number | Percent of AT (%) |
|---|---|---|---|---|---|---|
|
|
|
|
| 15,957 |
| 71.50 |
|
| 17,159 |
| 66.80 | |||
|
| 17,224 |
| 67.00 | |||
|
| 16,926 |
| 65.50 | |||
|
| 16,888 |
| 66.46 | |||
|
| 17,064 |
| 66.41 | |||
|
|
| 16,535 |
| 62.70 | ||
|
| 16,784 |
| 64.84 | |||
|
|
|
| 15,381 |
| 64.40 | |
|
| 15,469 |
| 63.70 | |||
|
| 15,585 |
| 63.70 | |||
|
| 16,128 |
| 63.80 | |||
| 19,507 |
| 64.00 | ||||
|
| 18,244 |
| 66.40 | |||
|
|
|
|
| 16,749 |
| 60.10 |
| Gene |
|
| ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Size (bp) | Start | End | Codon start/stop | Intergenic nucleotide (bp) | Size (bp) | Start | End | Codon start/stop | Intergenic nucleotide (bp) | |
|
| 1120 | 13 | 1132 | TTG/TAG | 36 | 1146 | 9247 | 10392 | ATG/TAA | 12 |
|
| 227 | 1169 | 1395 | TTG/T-- | 265 | 531 | 10405 | 10935 | ATG/TAG | -30 |
|
| 1661 | 2957 | -35 | 10906 | 12201 | |||||
|
| 700 | 2923 | 3622 | ATG/TAA | 23 | 699 | 12164 | 12862 | ATG/TAA | 15 |
|
| 3646 | 3713 | 76 | 12878 | 12943 | 77 | ||||
|
| 3790 | 4637 | -2 | 13021 | 13869 | -2 | ||||
|
| 790 | 4636 | 5425 | ATG/TAG | -2 | 789 | 13868 | 14656 | ATG/TAG | -1 |
|
| 5424 | 5491 | 5 | 14656 | 14722 | 6 | ||||
|
| 899 | 5497 | 6395 | GTG/T-- | 1639 | 898 | 14729 | 15626 | ATT/T-- | 443 |
|
| 8035 | 8102 | 48 | 113 | 179 | 45 | ||||
|
| 725 | 8151 | 8875 | ATG/T-- | 132 | 726 | 225 | 950 | ATG/T-- | 256 |
|
| 9008 | 9072 | -20 | 1207 | 1270 | 7 | ||||
|
| 287 | 9053 | 9339 | ATT/T-- | 33 | 288 | 1278 | 1565 | ATG/TAA | 1 |
|
| 9373 | 9439 | 20 | 1567 | 1632 | 10 | ||||
|
| 9460 | 9525 | 122 | 1643 | 1707 | 122 | ||||
|
| 1354 | 9648 | 11001 | TTG/TAG | 8 | 1254 | 1830 | 3083 | TTG/T-- | 103 |
|
| 11010 | 11076 | -1 | 3187 | 3250 | 2 | ||||
|
| 11076 | 11144 | 2 | 3253 | 3319 | 3 | ||||
|
| 11147 | 11213 | 13 | 3323 | 3387 | 18 | ||||
|
| 11227 | 11294 | -7 | 3406 | 3472 | -6 | ||||
|
| 11288 | 11351 | 30 | 3467 | 3529 | 33 | ||||
|
| 337 | 11382 | 11718 | ATA/TAA | 15 | 333 | 3563 | 3895 | ATT/TAG | -1 |
|
| 11734 | 11800 | 9 | 3895 | 3960 | 3 | ||||
|
| 11810 | 11876 | 8 | 3964 | 4029 | 15 | ||||
|
| 11885 | 11951 | -1 | 4045 | 4110 | 0 | ||||
|
| 11951 | 12018 | 5 | 4111 | 4178 | 10 | ||||
|
| 12024 | 12092 | 42 | 4189 | 4253 | 2 | ||||
|
| 12135 | 12200 | 23 | 4256 | 4320 | 5 | ||||
|
| 12224 | 12288 | 223 | 4326 | 4389 | 200 | ||||
|
| 1488 | 12512 | 13999 | CGA/ | 142 | 1512 | 4590 | 6101 | ATT/T-- | 154 |
|
| 14142 | 14209 | 8 | 6256 | 6320 | 0 | ||||
|
| 919 | 14218 | 15136 | GTG/TAA | 2 | 927 | 6321 | 7247 | ATG/TAA | 4 |
|
| 15139 | 15207 | 1 | 7252 | 7319 | 0 | ||||
|
| 15209 | 15274 | 2 | 7320 | 7383 | 0 | ||||
|
| 15277 | 15343 | 35 | 7384 | 7449 | 36 | ||||
|
| 1443 | 15379 | 16821 | ATT/T-- | 350 | 1441 | 7486 | 8926 | ATT/ | 320 |
| Species | AT (%) | GC (%) | AT skew | GC skew |
|---|---|---|---|---|
|
| 67.00% | 32.90 | -0.22687 | 0.367781 |
|
| 66.80% | 28.50 | -0.21958 | 0.319298 |
|
| 71.50% | 33.20 | -0.23653 | 0.379518 |
| Gene | lnL0 | lnL1 | Np0 | Np1 | Omega | P value | Positively selected sites (PSGs) |
|---|---|---|---|---|---|---|---|
|
| -1552.22 | -1552.22 | 17 | 18 | 2.52856 | 1 | |
|
| -4134.56 | -4134.56 | 17 | 18 | 2.35774 | 1 | |
|
| -4826.79 | -4826.79 | 17 | 18 | 2.62875 | 1 | |
|
| -1236.77 | -1236.78 | 17 | 18 | 3.31711 | 0.895254 | |
|
| -6913.41 | -6842.58 | 17 | 18 | 3.34388 | 0 | 140 A 0.509, 143 F 0.547, 144 L 0.865, 442 A 0.700, 446 F 0.620 |
|
| -5205.94 | -5205.94 | 17 | 18 | 2.64645 | 0.998872 | |
|
| -4693.2 | -4693.2 | 17 | 18 | 3.07094 | 1 | |
|
| -877.007 | -877.007 | 17 | 18 | 2.68959 | 1 | |
|
| -6088.71 | -6088.71 | 17 | 18 | 3.25046 | 1 | |
|
| -3173.94 | -3173.94 | 17 | 18 | 2.39696 | 1 | |
|
| -2763.71 | -2763.71 | 17 | 18 | 1.96195 | 1 | |
|
| -2882.5 | -2884.28 | 17 | 18 | 3.30394 | 0.058789 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAquatic Invertebrate Ecology and Behavior · Marine Biology and Ecology Research · Genomics and Phylogenetic Studies
Introduction
Pharidae belongs to Solenoidea which is one of the most ecologically and commercially significant superfamilies of marine Bivalvia, and the North-West and Indo-West Pacific regions exhibit the highest levels of species diversity, encompassing approximately 85% of all species, predominantly distributed in intertidal zones (Lin 2009; Saeedi et al. 2016; Costello and Saeedi 2019). According to the China Fisheries Statistics Yearbook (2024), the annual output of razor clams is 850,000 tons, accounting for 5.16% of the total output of mollusks. This Pharidae family has an extensive fossil record, dating back to approximately 103 million years ago (Mya) in the middle Cretaceous (Bolotov et al. 2018b). Although Pharidae is well-established as a clade, the internal taxonomic research has been ongoing and remains a contentious problem. Cosel (1993) promoted Solenidae to Solenoidea in 1993 and divided the superfamily into Solenidae and Pharidae according to the number of main teeth. Among them, the genus Sinonovacula was once classified by Graham into the family Solecurtidae, which belongs to the superfamily Tellinoidea (Graham 1935). However, an increasing number research findings contradict this, where the genus Sinonovacula should be categorized into the family Pharidae (Taylor et al. 2007; Guoquan et al. 2010; Yuan et al. 2012c; Yu et al. 2016). For example, the comparison of mitogenomes of six heterodont bivalves indicated that S.constricta (Lamarck, 1818) was more closely related to Solengrandis (Dunker, 1862), which belonged to Solenidae (Yuan et al. 2012c). The phylogenetic tree and molecular clock of tandem mitochondrial gene and nuclear gene (COI, 16S, 28S) revealed that Siliqua, Sinonovacula, Cultellus, and Novaculina belonged to Pharellinae, and Pharellajavanica (Lamarck, 1818) was classified under the Sinonovacula subclade (Bolotov et al. 2018b). Moreover, Pharidae were divided into four subfamilies which were composed of 14 existing genera, including Pharinae (Nasopharus, Pharus, Sinupharus), Cultellinae (Afrophaxas, Cultellus, Ensis, Ensiculus, Phaxas, Sinucultellus), Siliquinae (Siliqua), and Pharellinae (Novaculina, Orbicularia, Pharella, Sinonovacula), and Bolotov et al. (2018b) argued that Novaculininae was considered to be a junior synonym of Pharellinae (Appeltans et al. 2012; Signorelli et al. 2021). Nevertheless, since the above studies are only based on a limited number of taxa, the phylogenetic relationship of Pharidae has not been fully studied.
Mitochondrial DNA (mtDNA) is a genetic material independent of the nucleus DNA. Owing to their small size, rapid evolution, maternal inheritance, and simple structure, mitogenomes have become an attractive candidate tool for resolving phylogenetic relationships across a wide spectrum of metazoans (Boore 1999; Curole and Kocher 1999; Saccone et al. 1999; Miya et al. 2001; Gissi et al. 2008; Osigus et al. 2013; Cameron 2014). Mitogenomes of metazoan are usually circular double-stranded molecules, and range in size from 14 kb to 42 kb (Okimoto et al. 1992; Wolstenholme 1992; Smith and Snyder 2007). The typical mitogenome is composed of 37 genes compactly organized in a near-invariant arrangement, including 13 protein-coding genes of the oxidative phosphorylation (OXPHOS) system (cox1–3, cob, nad1–6, nad4L, atp6, atp8), 22 transfer RNAs (tRNAs) and two ribosomal RNAs homologous to the 16s and 23s of Escherichiacoli (rrnS and rrnL) (Wolstenholme 1992; Shadel and Clayton 1997; Andrews et al. 1999; Boore 1999). In general, metazoan mtDNA molecules have few or no nucleotides between genes except for a single non-coding region that contains signals for regulating replication and transcription (designated as the control region) (Clayton 1984; Wolstenholme 1992; Shadel and Clayton 1997). However, the phylum Mollusca has generated a vast array of unexpected deviations from the ‘textbook description’, including exceptional variation in size, frequent genome rearrangements, the integration of novel genes, and a complex inheritance system dubbed ‘doubly uniparental inheritance’ (Wu et al. 2012; Williams et al. 2017; Wu et al. 2019; Malkócs et al. 2022).
In mollusks, with the development of DNA sequencing technology, a large number of mitogenomes have been determined during the last thirty years (Yokobori et al. 2004; Yuan et al. 2012d; Kong et al. 2020; Ma et al. 2023; Taite et al. 2023). For instance, through comparing the complete mtDNA sequences of three scallop species from the subfamily Chlamydinae, it was found that the three genomes exhibited high variation in non-coding regions and different tRNA gene sets (Wu et al. 2009). Besides, the results of the phylogenetic analysis based on concatenated 12 protein-coding genes (PCGs) and two rRNA genes validated the monophyly of the family Mactridae and indicated that genera Mactrinula, Spisula, Rangia, and Mulinia should not be placed under subfamily Mactrinae (Ma et al. 2023). Nevertheless, to date, only four mitogenomes of Pharidae, which are ecologically and economically important deep-burrowing bivalves, are available (Zheng et al. 2010; Feng et al. 2021; Li et al. 2022).
Sinonovacularivularis (R. Huang & Y.-F. Zhang, 2007), the member of the genus Sinonovacula, is similar to S.constricta in reproduction and morphology (Huang and Zhang 2007). In contrast to S.constricta, which exhibits tolerance to wide salinity (5–40 ppt), S.rivularis is capable of thriving in low salt aquatic environments (4–20 ppt), and can even endure in freshwater conditions for over four days (Huang and Zhang 2007; Wang et al. 2009; Peng et al. 2019; Wang et al. 2024). In addition, a typical freshwater genus Novaculina is found in the family Pharidae (Schram 2010; Bolotov et al. 2018b). As a species of Novaculina, N.chinensis (Y.-Y. Liu & W.-Z. Zhang, 1979) was first discovered in Taihu Lake and Gaoyou Lake in China (Liu 1979). However, due to the pollution of water and the lack of protection awareness, they have been in danger of extinction (Liu 1979; Rao et al. 2003). In this study, we assembled the complete mitogenome of S.rivularis and N.chinensis, and analyzed their basic genome characteristics, nucleotide composition and relative synonymous codon usage (RSCU). The phylogenetic tree of Solenoidea was constructed and gene arrangement events between Pharidae and Solenidae were predicted. Furthermore, selective pressure analysis was conducted to explore the evolutionary adaptation of freshwater and marine species. Briefly, our findings will enrich the basis for the taxonomic study of Pharidae and contribute to deepening the understanding of the phylogenetic relationship between Solenoidea and its related groups.
Materials and methods
Sample collection
The samples for whole-genome sequencing of S.rivularis and N.chinensis were collected from the coastal area of Quanzhou in Fujian Province and the Qiantang River in Zhejiang Province, respectively, following the relevant guidelines and regulations. A total of ten individuals each of S.rivularis and N.chinensis were sampled, with average shell length of 55.98 ± 3.47 mm and 45.41 ± 2.74 mm, respectively. All specimens were preserved in 85% ethanol as voucher specimens. These specimens were deposited at Zhejiang Key Laboratory of Aquatic Germplasm Resource, Zhejiang Wanli University, Ningbo, China.
Mitogenome assembly and annotation
Raw genome reads were acquired through both Illumina HiSeq sequencing and PacBio Sequel IIe third-generation sequencing (unpublished), and assembled for the mitogenomes of these two species. Initially, a de novo mitogenome assembly was carried out with SPAdes v3.9.0 after filtering the unqualified reads by Trimmomatic v. 0.39 (Bankevich et al. 2012; Bolger et al. 2014). The scaffold sequences were then obtained by extending the contigs using SSPACE. The assembly quality was evaluated by GetOrganelle software (Jin et al. 2020). Finally, the MitoZ program was used to annotate the protein-coding genes (PCGs), two ribosomal RNAs (rRNAs) and transfer RNAs (tRNAs) (Meng et al. 2019).
Mitogenome characteristics analysis
The content and proportion of nucleotide bases were analyzed by MEGA 11. The base skew values were calculated according to the formulae: AT-skew = (A − T) / (A + T) and GC-skew = (G − C) / (G + C). The RSCU of the two mitogenomes was counted using PhyloSuite v1.2.3.
Phylogenetic analysis and gene arrangement analysis
To explore the evolutionary relationship of S.rivularis and N.chinensis, the published mitogenome sequences of Solenoidea and Hiatelloidea were retrieved from GenBank, and Solecurtusdivaricatus was selected as the outgroup (Table 1). The phylogenetic analysis was performed using PhyloSuite software (Zhang et al. 2020). First, using an invertebrate mitochondrial code table, MAFFT was used to independently align 12 protein-coding genes. The ATP8 gene was excluded due to its deletion in the majority of mollusks. Poorly aligned regions of the sequences were pruned by Gblocks under default parameters. The resulting alignments were then concatenated and transferred to ModelFinder for the best model prediction. Phylogenetic trees were estimated through maximum likelihood (ML) and Bayesian inference (BI) methods. The ML phylogenetic tree was generated using IQ-Tree with 1000 bootstrap replicates. The BI analyses were performed by MrBayes 3.2.6 with Markov Chain Monte Carlo (MCMC) for 5000,000 generations. The first 25% of trees were discarded as burn-in and the sampling was terminated when the convergence value was less than 0.01. The iTOL tool was exploited to visualize the phylogenetic tree (https://itol.embl.de/).
In addition, the most plausible gene order rearrangement events that might have occurred between Pharidae and Solenidae were reconstructed by pairwise comparisons of mitogenomes through the Common Interval Rearrangement Explorer (CREx) (Bernt et al. 2007).
Selective pressure analysis
The branch-site model was used to analyze the selection pressure on 12 PCGs of razor clams in the PAML package. In this model, N.chinensis was marked as the foreground branch to investigate the evolutionary adaptation between freshwater and marine species. The null model (model = 2, Nssites = 2, fix_omega = 1, omega = 1) and alternative model (model = 2, Nssites = 2, fix_omega = 0, omega = 2) were compared by likelihood ratio test (LRT). Subsequently, P-values were calculated through the chi-square distribution. Then, the posterior probability of the amino acid sites under positive selection was calculated according to the Bayesian empirical Bayes (BEB) method. The inference of positively selected sites was based on a posterior probability of greater than 95%.
Results
General features of S.rivularis and N.chinensis mitogenomes
The lengths of S.rivularis and N.chinensis mitogenomes were 17,159 bp and 15,957 bp, respectively (Fig. 1A). Both mitogenomes contain 12 PCGs, 22 tRNAs, and 2 rRNAs, all of which were located on the heavy chain. The ATP8 gene was missing in this two mitogenomes. Their composition was similar to that of other species in Pharidae, indicating a certain degree of conservation in this family. The detailed genes information was shown in Table 2. The base composition of S.rivularis and N.chinensis mitogenomes was displayed in Table 3 with AT contents of 66.80% and 71.50%, respectively, both of which exhibited an obvious AT bias. The AT content of N.chinensis was the highest among the published Adapedonta mitogenomes. In addition, the two mitogenomes all exhibited negative AT-skew and positive GC-skew, reflecting that the base composition ratios were A biased to T, and G biased to C. There were some differences in the types of start and termination codons of 12 PCGs between the two species (Table 2). Specifically, the start codons of 12 genes in S.rivularis were found to be ATN, TTG and GTG types, whereas in N.chinensis, all genes began with the codon ATN, with the exception of the ND4 gene, which used TTG as the start codon. Concerning termination codons, six genes in S.rivularis (cytb, atp6, cox3, nad4, nad3, nad1) and seven genes in N.chinensis (cytb, nad6, atp6, cox3, nad4l, nad3, nad1) were detected TAA or TAG at the sequence end. The remaining genes featured an incomplete termination codon consisting of a T that might be complemented into a complete stop codon by polyadenylation following transcription to the resultant mRNA (Ojala et al. 1981). Furthermore, the non-coding regions of the mitochondrial genomes of N.chinensis and S.rivularis account for 11.92% and 19.33%, respectively. The longest non-coding region (NCR) of N.chinensis and S.rivularis was both located between nad2 and trnK, with lengths of 443 bp and 1,639 bp respectively, which was identified as a putative control region (CR).
Maps of A the mitogenomes of S.rivularis and N.chinensis and their BRSCU.
As illustrated in Fig. 1B, the preferred codons for 22 amino acids of two species ended in A or U, consisting with the result of AT bias of the mitogenome sequence. As a consequence of the duplication of tRNA-Leu and tRNA-Ser, Leu and Ser were each encoded by six and eight codons, respectively. The most frequently used codons were UUA (Leu2), UCU (Ser2), GCU (Ala) and CCU (Pro). Compared to S.rivularis, CUG (Leu1), AUC (Ile), AAC(Asn) were utilized to a lesser extent in N.chinensis.
Phylogenetic analysis
The 12 protein-coding genes from 15 taxa were concatenated to generate a sequence matrix of 10,806 bp. The tree topologies derived from the ML and BI analyses were largely congruent exhibiting high posterior probabilities (PP) and bootstrap support values (BS) in most nodes (Fig. 2). Phylogenetic analyses revealed that the genus Hiatella from Hiatelloidea was closely related to the superfamily Solenoidea, indicating a close evolutionary relationship between them. Additionally, both analyses strongly confirmed the monophyly of Solenoidea, which was divided into two major branches, Solenidae and Pharidae. In the family Pharidae, the genus Sinonovacula (including S.rivularis and S.constricta) was clustered alongside N.chinensis, with Cultellusattenuatus emerging as a sister group. Siliquaminima and Ensisleei were clustered in a separate cluster.
The phylogenetic trees based on concatenated 12 mitochondrial PCGs, and the gene orders of Adapedonta species. Values shown next to nodes are posterior probabilities (left) and ML bootstrap support values (right). Newly assembly mitogenomes are marked with triangles. Except for Panopeaabrupta (https://inverts.wallawalla.edu) and Panopeaglobosa (Góngora-Gómez et al. 2016), the images of the other species are all from https://www.inaturalist.org.
Gene arrangement
The mitogenomes of Solenoidea all exhibited the identical composition of 12 PCGs, 22 tRNAs, and 2 rRNAs, except for Ensisleei, which contained an additional ATP8 gene (Fig. 3). The gene arrangement was consistent within each family, and there was a certain level of conservation in gene arrangement between Solenidae and Pharidae. A large block, rrnL-ATP6-M-rrnS-cox3, and five small blocks, L2-nad1-L1, S-nad2, nad5-cytb, I-D, Q-C were shared by both families, providing further evidence of the close lineage relationship observed in the phylogenetic analysis of this study. The CREx analysis suggested that three transposition and four tandem duplication random losses (TDRLs) might have occurred between Pharidae and Solenidae.
Putative gene rearrangement events between Pharidae and Solenidae. Green and red lines represent transposition and TDRL events, respectively, which were step by step identified by CREx.
Select pressure analysis
The species of Solenoidea were selected for molecular evolution analysis, with N.chinensis designated as the foreground branch (Fig. 4). The branch-site model (BSM) in the PAML package was employed to detect positively selected genes (PSGs). As illustrated in Table 4, the substitution model A was significantly better than the neutral selection model null in nad5, indicating that this gene underwent positive selection in the foreground branch (P < 0.05). According to the BEB analysis, there were five positive selection sites in the nad5 amino acid sequences (140 A 0.509, 143 F 0.547, 144 L 0.865, 442 A 0.700, 446 F 0.620). Moreover, discrepancies were observed in the 144^th^ site between freshwater N.chinensis (Ala) and marine razor clams (Leu) (Fig. 5). However, the evidence for each site was somewhat inconclusive. These findings suggest that the nad5 gene may have played a pivotal role in the adaptive evolution of freshwater environments.
Phylogenetic tree of Solenoidea for selective stress analysis. The branch marked in red is the foreground branch.
The difference of the 144th positive selected amino acid site in NAD5 of eight Solenoidea species. The 144th site is indicated by a red frame.
Discussion
General features of Pharidae mitogenomes
The mitogenomes of S.rivularis and N.chinensis were newly assembled, with lengths of 17,159 and 15,957 bp, respectively. In compared with the previously sequenced AdapedontamtDNA size (ranged from 15,381 bp to 19,507 bp), their mitogenome sizes were within the normal range (Zheng et al. 2010; Yuan et al. 2012b; Feng et al. 2021; Li et al. 2022). Notably, the genome size of N.chinensis was the smallest in the family Pharidae, which was associated with the variation in length of the control region. The CR is the region with the largest sequence and length variation in the mitogenome, and has the fastest evolution, which is crucial for the regulation of mitochondrial DNA replication and transcription (Wolstenholme 1992; Boore 1999). The substantial differences in the content and structure of the control region within the mollusk lineage provide valuable insights for population genetic analysis (Sasuga et al. 1999; Tomita et al. 2002; Kawashima et al. 2013). Among the published mitogenomes of Pharidae, there is a large control region between nad2 and trnK, such as S.constricta (1,492 bp), S.minima (1,371 bp), C.attenuatus (1,173 bp) and E.leei (1,101 bp) (Zheng et al. 2010; Feng et al. 2021; Li et al. 2022). In this study, S.rivularis displayed a moderately larger control region size of 1,639 bp, whereas it was only 441 bp in N.chinensis, making it a different mitogenome size in the family Pharidae. Intriguingly, a similar control region was not observed in the species of Solenidae (Yuan et al. 2012a, b). This distinction provides evidence for the taxonomic division of the subfamily of Solenoidea.
Molecular phylogeny and gene arrangement of the family Pharidae
The topological tree constructed from the 12 mitochondrial PCGs sequence based on the BI and ML methods yielded consistent results, demonstrating that Solenoidea is clearly divided into Solenidae and Pharidae, which is consistent with the prior research results (Yuan et al. 2012d; Feng et al. 2021). Previously, S.rivularis was identified as a new species of Sinonovacula distinct from *S.constricta
- based on morphological studies and a comparative analysis of COI and 16SrRNA fragments (Huang and Zhang 2007; Weng et al. 2013). In this research, this classification view was supported at the level of mitogenomes, and Sinonovacula belonged to the family Pharidae (Adapedonta: Solenoidea). In addition, N.chinensis was previously classified into Solecurtidae, whereas the results of this study demonstrated that N.chinensis and Sinonovacula are clustered together, forming a novel branch in the family Pharidae, which was consistent with the taxa in WoRMS (Liu 1979; Appeltans et al. 2012). Recently, the phylogenetic tree and molecular clock of tandem mitochondrial gene and nuclear gene (COI, 16S, 28S) revealed that Siliqua, Sinonovacula, Cultellus, and Novaculina belonged to Pharellinae (Bolotov et al. 2018b). However, Cultellus and Siliqua were categorized into the subfamily Cultellinae and Siliquinae, respectively, by Ahyong (Appeltans et al. 2012). Previously, Pharidae were divided into four subfamilies: Pharinae (Nasopharus, Pharus, Sinupharus), Cultellinae (Afrophaxas, Cultellus, Ensis, Ensiculus, Phaxas, Sinucultellus), Siliquinae (Siliqua), and Pharellinae (Novaculina, Orbicularia, Pharella, Sinonovacula). However, the present results indicate that Pharidae are divided into two clades, in which Cultellus is clustered alongside the Sinonovacula and Novaculina, while Siliqua and Ensis clustered together. These observations reflect that the current categorization of the subfamily Pharidae requires further research and refinement, particularly in combination with more species information.
Unlike stable gene arrangements of Vertebrata and Arthropoda, the gene orders of all genes within mtDNA exhibit considerable variability in every major molluscan lineage, including Cephalopoda, Bivalvia, Scaphopoda, and Monoplacophora (Rawlings et al. 2001; Dreyer and Steiner 2004; Yuan et al. 2012c; Stöger et al. 2016; Ma et al. 2023). Gene rearrangements may be caused by reverse transpositions, transpositions, inversions, and TDRL, which can provide important clues about the evolutionary history of species (Boore and Brown 1998; Serb and Lydeard 2003; Wang et al. 2021). In this paper, CREx analysis predicted that three transpositions and four TDRLs might have occurred between Pharidae and Solenidae, implying that dramatic mitogenome changes occurred during species differentiation. Moreover, the gene order illustration of Adapedonta revealed that species with a closer genetic relationship tended to share a similar gene arrangement, indicating that there is a potential relationship between evolution and gene rearrangement (Fig. 2). However, in this study, three distinct gene arrangement types were observed in the family Hiatellidae, especially in the genus Hiatella with nad3 and nad1 transpositions in terms of 12 PCGs arrangement (Fig. 2). The similar case that different gene arrangements in the same genus has also been reported in the genera Dendropoma and Crassostrea (Rawlings et al. 2010; Ren et al. 2010). Therefore, the taxonomic evolution of species cannot be substantiated exclusively through the examination of gene sequences; it also necessitates the integration of phylogenetic reconstruction.
Adaptive evolution of Pharidae mitochondrial genes to freshwater environment
Pharidae is a major marine family, with the exception of Novaculina, that is a relict marine-derived freshwater lineage (Annandale 1922; Bolotov et al. 2018a). The branch-site model study was used to determine whether positive selection occurs at a few places in freshwater razor clam. The results suggested that the nad5 gene underwent positive selection. NADH dehydrogenase is the initial and most substantial enzyme complex in the respiratory chain, functioning as a proton pump (da Fonseca et al. 2008). Nad2, nad4, and nad5 are considered to be the actual proton pumping devices because of their sequence homology with a class of Na^+^/H^+^ antiporters (Brandt 2006). The efficiency of the proton transfer process may be interfered by the mutation of the complex, which could be a crucial factor in adaptive evolution (Hassanin et al. 2009; Yu et al. 2011). For instance, the outcomes of positive selection sites in mussels from disparate habitats reflected that the p-value of nad4 was significant in freshwater branches and six sites were identified as positive sites with BEB analysis (> 95%), which implies that nad4 may contribute to the adaptation of Limnopernafortunei in freshwater (Zhao et al. 2022). Significant non-synonymous changes were detected in the cytb and nad5 genes by comparing mitogenomes of panpulmonate gastropods that are distributed from marine to intertidal and terrestrial habitats (Romero et al. 2016). Therefore, the positive selection of nad5 gene in N.chinensis may be the result of the adaptive evolution of freshwater environment. Moreover, divergent selection occurred at site 144 of nad5, where the amino acids Ala and Leu were identified in the freshwater Novaculinachinensis and seven marine lineages, respectively, indicating divergent evolution exists the family Pharidae. Divergent evolution is the process by which separate species with common ancestors evolve distinct features to adapt to their unique living environment, which is one of the important mechanisms for the formation of biodiversity (Gautam 2020). However, the evidence supporting the positive selection of individual nad5 sites is insufficient. To provide more robust statistical support for the differences in evolutionary adaptation between freshwater and seawater species, it is necessary to include more freshwater razor clam sequences.
Conclusions
In summary, the mitogenomes of S.rivularis and N.chinensis were assembled using next-generation sequencing data, with the genomes measuring 17,159 bp and 15,957 bp, respectively. Both genomes consist of 12 protein-coding genes, 22 transfer RNA genes, and two ribosomal RNA genes. Among the published Pharidae mitogenomes, N.chinensis exhibits the smallest genome size but the highest AT content. The results of the phylogenetic analysis showed that N.chinensis and Sinonovacula (S.constricta + S.rivularis) were closely related and belonged to the family Pharidae. The gene order rearrangements in Solenoidea can be attributed to transposition and TDRL events. Moreover, the nad5 genes carry a signal of positive selections in the foreground N.chinensis, which promotes the adaptation to freshwater environments. We also show that divergent evolution occurred at site 144 in the freshwater and marine lineages. Overall, this study provides further theoretical support for the phylogenetic relationship of Pharidae, and contributes to deepening the understanding of the mitogenomic adaptations of Pharidae.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Andrews RM Kubacka I Chinnery PF Lightowlers RN Turnbull DM Howell N (1999) Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA.Nature Genetics 23(2): 147–147. 10.1038/1377910508508 · doi ↗ · pubmed ↗
- 2Annandale N (1922) The Marine Element in the Fauna of the Ganges. Bijdr Dierk Amsterdam, 143–154. 10.1163/26660644-02201020 · doi ↗
- 3Appeltans W Ahyong S Anderson G Angel M Artois T Bailly N Bamber R Barber A Bartsch I Berta ABłażewicz-Paszkowycz M Bock P Boxshall G Boyko C Brandão S Bray R Bruce N Cairns S Chan TY Cheng L Collins A Cribb T Curini-Galletti M Dahdouh-Guebas F Davie P Dawson M De Clerck O Decock W De Grave S De Voogd N Domning D Emig C Erséus C Eschmeyer W Fauchald K Fautin D Feist S Fransen C Furuya H Garcia-Alvarez O Gerken S Gibson D Gittenberger A Gofas SGómez-Daglio L Gordon D Guiry M Hernandez F Hoeksema B Hopcroft R Jaume D Kirk P Koedam N Koenemann S Kolb J Kristensen R Kroh A Lambert G Lazarus D Lemaitre R Longshaw M Lo · doi ↗ · pubmed ↗
- 4Bankevich A Nurk S Antipov D Gurevich AA Dvorkin M Kulikov AS Lesin VM Nikolenko SI Son P Prjibelski AD Pyshkin AV Sirotkin AV Vyahhi N Tesler G Alekseyev MA Pevzner PA (2012) SP Ades: a new genome assembly algorithm and its applications to single-cell sequencing.Journal of Computational Biology 19(5): 455–477. 10.1089/cmb.2012.002122506599 PMC 3342519 · doi ↗ · pubmed ↗
- 5Bernt M Merkle D Ramsch K Fritzsch G Perseke M Bernhard D Schlegel M Stadler PF Middendorf M (2007) CR Ex: inferring genomic rearrangements based on common intervals.Bioinformatics 23(21): 2957–2958. 10.1093/bioinformatics/btm 46817895271 · doi ↗ · pubmed ↗
- 6Bolger AM Lohse M Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data.Bioinformatics 30(15): 2114–2120. 10.1093/bioinformatics/btu 17024695404 PMC 4103590 · doi ↗ · pubmed ↗
- 7Bolotov IN Aksenova OV Bakken T Glasby CJ Gofarov MY Kondakov AV Konopleva ES Lopes-Lima M Lyubas AA Wang Y Bychkov AY Sokolova AM Tanmuangpak K Tumpeesuwan S Vikhrev IV Shyu JBH Win T Pokrovsky OS (2018 a) Discovery of a silicate rock-boring organism and macrobioerosion in fresh water.Nature Communications 9(1): 2882–2892. 10.1038/s 41467-018-05133-4PMC 605653230038289 · doi ↗ · pubmed ↗
- 8Bolotov IN Vikhrev IV Lopes-Lima M Lunn Z Chan N Win T Aksenova OV Gofarov MY Kondakov AV Konopleva ES Tumpeesuwan S (2018 b) Discovery of Novaculinamyanmarensis sp nov (Bivalvia: Pharidae: Pharellinae) closes the freshwater razor clams range disjunction in Southeast Asia.Scientific Reports 8(1): 16325–16336. 10.1038/s 41598-018-34491-830397264 PMC 6218526 · doi ↗ · pubmed ↗