[3+2] Cycloaddition to a Chiral 5-Methylene-1,3-dioxolan-4-one and Pyrolysis of the Spiro Adducts
R. Alan Aitken, Lynn A. Power, Alexandra M. Z. Slawin

TL;DR
This paper studies chemical reactions involving a chiral compound derived from lactic acid and explores the outcomes of pyrolysis of the resulting adducts.
Contribution
The study presents new [3+2] cycloaddition reactions and novel gas-phase pyrolysis pathways for a chiral dioxolanone derivative.
Findings
Spiro adducts were successfully formed with benzonitrile oxide, acetonitrile oxide, diazomethane, and diphenyldiazomethane.
Pyrolysis of the adducts led to diverse products, including pivalaldehyde, CO2, and benzophenone.
Failed attempts with nitrile sulfide, nitrile imine, and azides highlight selectivity in the reaction conditions.
Abstract
The [3+2] cycloaddition chemistry of (2S)-5-methylene-2-t-butyl-1,3-dioxolan-4-one, derived from lactic acid, has been examined, and spiro adducts have been obtained with benzonitrile oxide, acetonitrile oxide, diazomethane and diphenyldiazomethane. The structure and absolute stereochemistry of the benzonitrile oxide adduct has been confirmed by X-ray diffraction, and all the adducts have been fully characterised by 1H and 13C NMR. Attempted cycloaddition with a nitrile sulfide, a nitrile imine and azides failed. Pyrolysis results in a range of novel gas-phase reactions, with the nitrile oxide adducts giving pivalaldehyde, CO2, the nitrile and ketene, the diazomethane adduct losing only N2 to give a cyclopropane-fused dioxolanone, and the diphenylcyclopropane derived from diphenyldiazomethane giving mainly benzophenone in a sequence involving the loss of pivalaldehyde and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
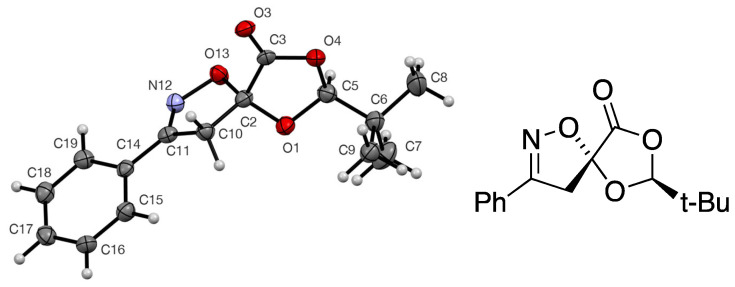 Figure 1
Figure 1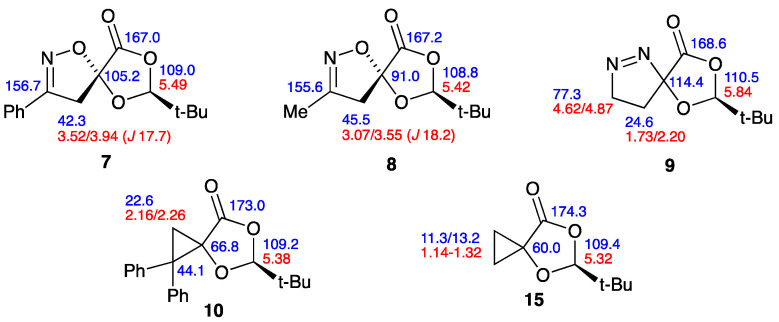 Figure 2
Figure 2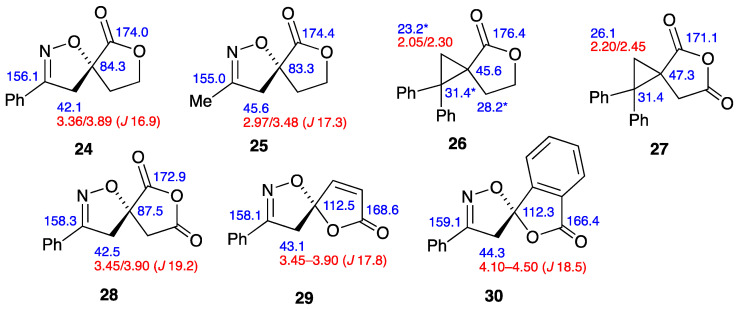 Figure 3
Figure 3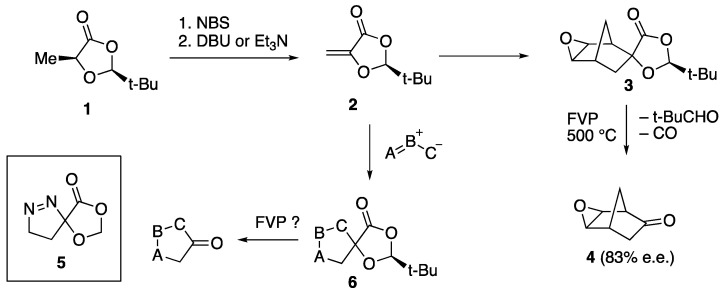 Figure 4
Figure 4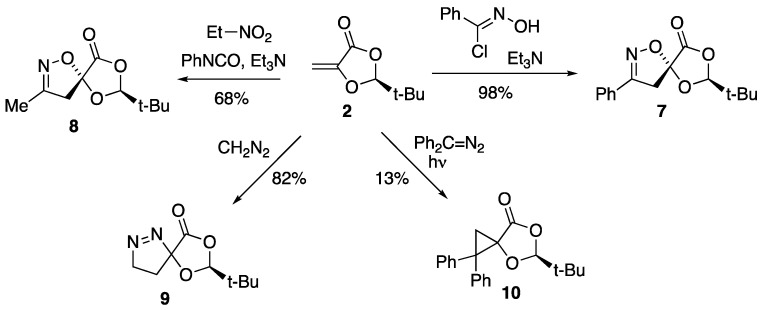 Figure 5
Figure 5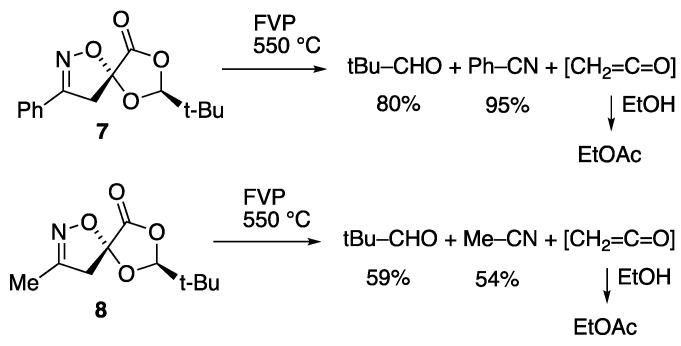 Figure 6
Figure 6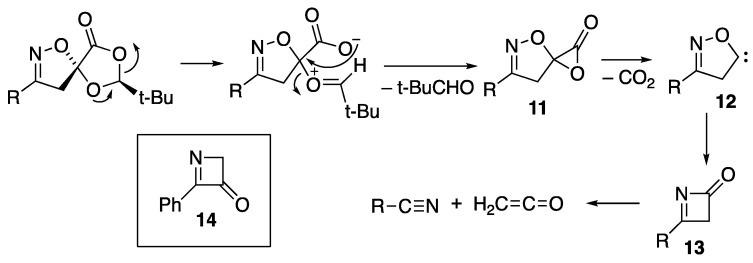 Figure 7
Figure 7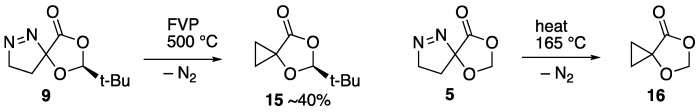 Figure 8
Figure 8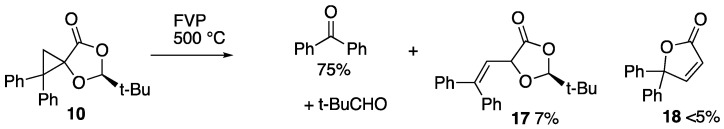 Figure 9
Figure 9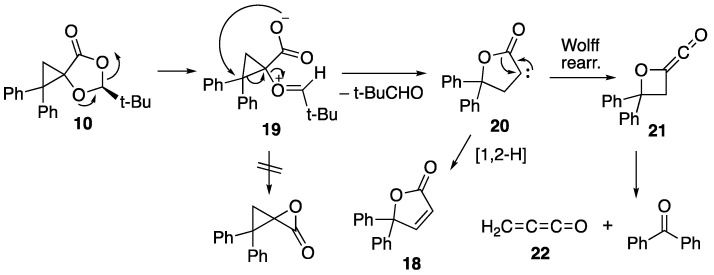 Figure 10
Figure 10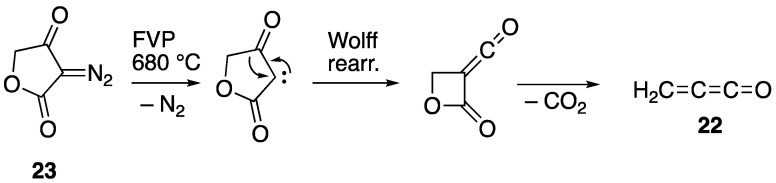 Figure 11
Figure 11- —School of Chemistry
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsOrganic Chemistry Cycloaddition Reactions · Cyclopropane Reaction Mechanisms · Oxidative Organic Chemistry Reactions
1. Introduction
Spiro heterocycles have been of considerable recent interest due to their rigidly defined geometry and potential as scaffolds in medicinal chemistry [1,2,3]. In previous work, it has been shown that chiral dioxolanones such as compound 1 (Scheme 1) readily derived from (S)-lactic acid have considerable value in a range of asymmetric synthesis methods [4,5]. Some time ago, we showed that deprotection by flash vacuum pyrolysis (FVP) resulted in the loss of pivalaldehyde and CO from substituted dioxolanones to give ketones [6], thus making the starting dioxolanone 1 a chiral acetyl anion equivalent [7]. Bromination of 1 followed by dehydrobromination affords the 5-methylene compound 2 [8], and this has been found to undergo a range of Diels–Alder reactions [9,10,11,12]. We recently described the Diels–Alder reaction of 2 with several 1,3-dienes, and again, deprotection of the adducts using FVP was found to afford ketones in some cases. This is illustrated by the reaction of epoxide 3, formed by the cycloaddition of 2 with cyclopentadiene followed by epoxidation, to give the epoxynorbornanone 4 in high e.e. [13]. Overall, compound 2 acts as a chiral ketene equivalent.
As far as we are aware, the [3+2] cycloaddition chemistry of chiral dioxolanone 2 has never been investigated, and indeed we are only aware of a single example of [3+2] cycloaddition to any 5-alkylidene-1,3-dioxolan-4-one, compound 5 formed from the 2-unsubstituted dioxolanone and diazomethane [14]. The historical development of the [3+2] cycloaddition reaction and its mechanistic understanding has been chronicled in a recent review [15]. Methylenedioxolanone 2 is a particularly interesting 2π-component for [3+2] cycloaddition, since the bulky tert-butyl group introduces a strong facial bias and the carbonyl and enol-ether functions exert opposing polarising effects on the C=C double bond. Based on previous studies of its [4+2] cycloaddition with both homo- and hetero-dienes [10], it is expected that addition will occur on the face remote from the tert-butyl and that the polarising effect of the enol ether will dominate, causing the “negative” end of any TAC to add at the ring carbon. In this paper, we describe the cycloaddition of 2 with a range of three-atom components (TACs, formerly known as 1,3-dipoles [16]) to give spiro heterocycles of general structure 6, and FVP of these adducts.
2. Results and Discussion
2.1. [3+2] Cycloaddition to Methylenedioxolanone 2
Treatment of compound 2 and benzhydroximoyl chloride in diethyl ether with triethylamine [17] at 0 °C for 30 min followed by room temperature for 30 min gave the crystalline benzonitrile oxide adduct 7 in almost quantitative yield as a single stereoisomer (Scheme 2). This showed the expected NMR signals for a diastereotopic CH_2_ group [δ_H_ 3.52, 3.94 (^2^J 17.7 Hz); δ_C_ 42.3], as well as a signal for the spiro carbon [δ_C_ 105.2]. The corresponding acetonitrile oxide adduct 8 was also readily prepared by addition of a solution of 2 and phenyl isocyanate in toluene to a cooled solution of nitroethane in toluene containing a catalytic quantity of triethylamine [18]. The product was isolated as an oil in moderate yield, again showing characteristic NMR signals for the diastereotopic CH_2_ group [δ_H_ 3.07, 3.55 (^2^J 18.2 Hz), δ_C_ 91.0], this time with additional four-bond coupling to the methyl group (^4^J 0.8 Hz) and a signal for the spiro carbon [δ_C_ 91.0]. Again, the compound was obtained as a single stereoisomer, and from the similarity of the NMR data it seemed clear that 7 and 8 were formed with the same regio- and stereo-selectivity.
The crystals of compound 7 were suitable for X-ray structure determination, and this gave the molecular structure shown (Figure 1, Table 1), in which the TAC has added to the face of the C=C double bond away from the bulky tert-butyl group and with the more electronegative end of the TAC adding to the ring carbon of the C=C double bond. It is thus clear that this 2-π component is exhibiting the same facial and regio-selectivity in [3+2] cycloaddition as expected from the earlier studies on its [4+2] cycloaddition with heterodienes [10]. There were no significant intermolecular interactions in the crystal structure. We assume that the acetonitrile oxide adduct 8 has the corresponding stereochemistry, as depicted in Scheme 2.
Reaction with related TACs was now examined, but the dioxolanone 2 failed to react with either benzonitrile sulfide (formed by thermolysis of 5-phenyl-1,3,4-oxathiazol-2-one [19]) or with diphenylnitrile imine (formed by triethylamine treatment of α-chlorobenzaldehyde phenylhydrazone [20]).
As mentioned earlier, the one literature example of [3+2] cycloaddition to a methylenedioxolanone is compound 5, formed from diazomethane [14]. Compound 2 was therefore treated with an ethereal solution of diazomethane, and the expected adduct 9 was formed in good yield. This showed distinctive NMR signals for two adjacent but strongly differentiated CH_2_ groups, corresponding to C–CH_2_ [δ_H_ 1.73, 2.20; δ_C_ 24.6] and =N–CH_2_ [δ_H_ 4.62, 4.87; δ_C_ 77.3], together with a signal for the spiro carbon [δ_C_ 114.4]. Compound 2 failed to react with diphenyldiazomethane under thermal conditions, but upon photolysis a new product was formed in low yield which turned out to be the cyclopropane 10. This showed an unexpectedly complex NMR pattern, with restricted rotation in the sterically hindered phenyl groups leading to separate signals for some of the normally equivalent aromatic carbons, [δ_C_ 6 × CH, 2 × 2CH].
The reactivity of dioxolanone 2 with ethyl diazoacetate, ethyl azidoformate and phenyl azide was also examined under both thermal and photochemical conditions, but the starting material was recovered unchanged in all cases. Perhaps owing to steric hindrance and the push–pull substituents, this seems to be a rather reluctant 2-π electron component.
2.2. Flash Vacuum Pyrolysis of the Spiro Cycloadducts
Having obtained the four spiro cycloadducts 7–10 with novel structures, we were interested to examine their thermal decomposition. Based on our previous work [13], this was initially expected to yield cyclic ketones 6 by loss of pivalaldehyde and carbon monoxide (Scheme 1).
Subjecting the benzonitrile oxide adduct 7 to FVP led to complete reaction at a temperature of 550 °C (Scheme 3), to give high yields of benzonitrile and pivalaldehyde recovered from the cold trap after warming to room temperature, identified by ^1^H and ^13^C NMR spectroscopy (see Supplementary Materials).
Mechanistic considerations led to the suspicion that ketene might also be produced, and this was confirmed by adding a small amount of ethanol to the trap while still cold, which led to the formation of ethyl acetate upon warming up. FVP of the acetonitrile oxide adduct 8 also led to complete reaction at the same temperature, giving acetonitrile, pivalaldehyde and ketene.
Based on our previous work [13], we believe that this process is initiated by ring opening to an oxonium carboxylate which then loses pivalaldehyde to form the spiro α-lactone 11 (Scheme 4). Rather than undergoing loss of CO to form an isoxazolinone, as observed, for example, in the conversion of 3 into 4, it is apparently more favourable for this adduct to lose CO_2_ to give the heterocyclic carbene 12, which readily undergoes oxa-carbene rearrangement, resulting in ring contraction to the 3H-azetin-2-one 13, which then fragments to the nitrile and ketene.
Although such azetinones are unknown, it is interesting to note that there is one claim to have generated the isomeric 4-phenyl-2H-azetin-3-one 14 during the photolysis of benzonitrile in the presence of 1,3-dioxole followed by subsequent hydrolysis and dehydration [21]. However, the data presented, including in particular a ^1^H NMR signal at δ_H_ 7.9 ppm attributed to CH_2_ with no corresponding signal apparent in the ^13^C NMR spectrum, do not support the structure shown.
We now turned to the diazomethane adduct 9, and it was found to undergo complete reaction upon FVP at 500 °C. However, rather than any process involving the dioxolanone, it was clear that there had simply been extrusion of N_2_ to give the spiro cyclopropane–dioxolanone 15 (Scheme 5). This was obtained in low yield as an oil and gave satisfactory HRMS data, as well as distinctive low-frequency signals for the two non-equivalent CH_2_ groups in its NMR spectra [δ_H_ 1.14–1.32; δ_C_ 11.3, 13.2] and a signal for the spiro carbon [δ_C_ 60.0]. This behaviour is in good agreement with that observed for the 2-unsubstituted analogue 5, which was reported to lose N_2_ to afford 16 upon being slowly added to a flask containing pieces of porcelain heated at 165 °C [14].
Finally, the thermal reactivity of spiro diphenylcyclopropane–dioxolanone 10 was examined. Reaction was again complete upon FVP at a furnace temperature of 500 °C, and the NMR analysis of the product showed the presence of pivalaldehyde and several phenyl-containing compounds. The latter were separated by preparative TLC to give a main fraction which crystallised and was found to consist mainly of benzophenone (75%), together with a little starting material and a new compound in low yield which we believe to be the diphenylvinyldioxolanone 17, isomeric with 10 (Scheme 6). This exhibited distinctive mutually coupled doublets in the ^1^H NMR spectrum [δ_H_ 5.12, 6.12 (^3^J 10 Hz)] as well as the expected 1H and 9H singlets but, due to the low yield, further characterisation was not possible. A second, less polar fraction gave distinctive signals for 4,4-diphenylbutenolide 18 [22], but this was only formed in less than 5% yield.
While the minor product 17 is a result of simple thermal ring-opening of the cyclopropane, we propose that both the major product benzophenone and 18 are formed by the mechanism shown in Scheme 7.
In this scheme, the normal fragmentation initially gives the oxonium carboxylate 19 but, rather than cyclising with loss of pivalaldehyde to give the extremely strained α-lactone, as happens for the nitrile oxide adducts 7 and 8, this rearranges by attack of the carboxylate on the electron-poor diphenylcyclopropane carbon, resulting in loss of pivalaldehyde and formation of the butyrolactone carbene 20. As a minor process, this can undergo a 1,2-hydrogen shift to form the stable butenolide 18. However, it appears that it prefers to undergo Wolff rearrangement to give the oxetanylideneketene 21, which is set up to undergo cycloreversion to form benzophenone and the unstable methyleneketene (propadienone) 22. Compound 22, when generated using FVP, cannot be isolated under normal conditions and forms a glassy polymer [23], which we assume is its fate here. The formation of 22 in the present reaction is rather similar to the method used by Chapman to generate it for low-temperature matrix IR studies, starting from the diazo compound 23 (Scheme 8) [24].
2.3. NMR Data for the Spiro Compounds
In the course of this work, we have obtained products containing a range of novel spiro heterocyclic ring systems. For these, the ^1^H and ^13^C NMR data have been unambiguously assigned including, where necessary, using HSQC and HMBC methods. These data are combined and presented in pictorial form in Figure 2. While these data form a self-consistent pattern, it was realised that there are few closely similar spiro systems in the literature. We have therefore compiled the published data for the most closely similar systems, and this is presented in Figure 3.
As mentioned earlier, the only spiro dioxolanone product previously formed by [3+2] cycloaddition is compound 5, but due to the early date of its preparation there are no NMR data either for it nor its cyclopropane pyrolysis product 16. The most closely analogous structures for which there are NMR data are formed from α-methylenebutyrolactone. Thus, its benzonitrile oxide adduct 24 was reported in 1995 [25], while the acetonitrile oxide adduct 25 was reported in 2015 [26]. It can be seen that there is good agreement with the data for 7 and 8 in both cases.
Similarly, the diphenylcyclopropane compound 26 obtained from α-methylenebutyrolactone [27] provides a good comparison with the data for 10, although there is some ambiguity in the assignment of the marked signals, which were not assigned in the original publication. Further relevant spiro compounds are the diphenylcyclopropane 27 [28] and benzonitrile oxide adduct 28 [29], both derived from itaconic anhydride. Further, more diverse structures for comparison with the data of the benzonitrile oxide adduct 7 are 29, derived from “protoanemonin” [30], and 30, derived from 3-methylenephthalide [31]. Overall, we hope that this compilation of data may provide a useful reference for future workers in the field.
3. Experimental Procedure
3.1. General Experimental Details
NMR spectra were recorded for solutions in CDCl_3_ using Bruker instruments, and chemical shifts are given in ppm to high frequency from Me_4_Si with coupling constants J in Hz. On ^1^H NMR spectra, signals at 0.0 and 7.26 are due to internal Me_4_Si and residual CHCl_3_, respectively. The ^13^C NMR spectra are referenced to the solvent signal at 77.0. IR spectra were recorded on a Perkin Elmer 1420 instrument. Elemental analysis was conducted using a Carlo Erba CHNS analyser. Mass spectra were obtained using a Micromass instrument, and the ionisation method used is noted in each case. Column chromatography was carried out using silica gel of 40–63 μm particle size, and preparative TLC was carried out using 1.0 mm layers of Merck alumina 60G containing 0.5% Woelm fluorescent green indicator on glass plates. Melting points were recorded on a Gallenkamp 50W melting point apparatus or a Reichert hot-stage microscope. Optical rotation measurements were made using an Optical Activity 1000 polarimeter and are given in units of 10^−1^ deg cm^2^ g^−1^.
Flash vacuum pyrolysis (FVP) was carried out in a conventional flow system by subliming the starting material through a horizontal quartz tube (30 × 2.5 cm) externally heated by a tube furnace to 500–550 °C and maintained at a pressure of 2–5 × 10^−2^ Torr by a rotary vacuum pump. Products were collected in a liquid-N_2_-cooled U-shaped trap and purified as noted.
General organic and inorganic reagents and solvents were obtained from standard suppliers and used as received. Dry diethyl ether was prepared by storage over sodium wire. The starting materials (2R)-2-t-butyl-5-methylene-1,3-dioxolan-4-one 2 [8], benzhydroximoyl chloride [32], diazomethane [33] and diphenyldiazomethane [34] were prepared by the reported methods.
3.2. Preparation of [3+2] Cycloadducts of (2R)-2-t-Butyl-5-methylene-1,3-dioxolan-4-one 2
3.2.1. Preparation of (2S,5R)-Spiro[2-t-butyl-1,3-dioxolan-4-one-5,5′-4′,5′-dihydro-3′-phenylisoxazole] 7
A solution of 2 (1.50 g, 9 mmol) and benzhydroximoyl chloride (1.50 g, 9.6 mmol) was stirred in Et_2_O (50 cm^3^) at 0 °C, while a solution of triethylamine (1.01 g, 10 mmol) in Et_2_O (8 cm^3^) was added dropwise over 30 min. The mixture was stirred for a further 30 min at room temperature. The precipitate, triethylamine hydrochloride, was filtered off and the filtrate was washed with water. The ethereal layer was dried and evaporated to give a pale yellow solid. Recrystallisation using Et_2_O/CH_2_Cl_2_/hexane gave the pure product 7 (2.43 g, 98%) as colourless needles, mp 107–109 °C; [α]D +1.0 (c = 1.0, CH_2_Cl_2_); (Found: C, 65.5; H, 6.3; N, 5.2. C_15_H_17_NO_4_ requires C, 65.4; H, 6.2; N, 5.1%); ν_max_/cm^−1^ 1804 (C=O), 1349, 1256, 1198, 1187, 1177, 1065, 1011, 962 and 853; δ_H_ 1.00 (9H, s, t-Bu), 3.52 and 3.94 (2H, AB pattern, J 17.7, 4′-CH_2_), 5.49 (1H, s, CH-t-Bu), 7.41–7.49 (3H, m, Ph) and 7.66–7.72 (2H, m, Ph); δ_C_ 23.1 (3CH_3_, t-Bu), 34.3 (C, CMe_3_), 42.3 (4′-CH_2_), 105.2 (5/5′-C), 109.0 (2-CH), 127.0 (2CH, Ph), 127.8 (C, Ph C-1), 129.0 (2 CH, Ph), 131.1 (CH, Ph C-4), 156.7 (C=N) and 167.0 (C=O); m/z (ES^+^) 298.21, (M+Na^+^, 100%).
3.2.2. Preparation of (2S,5R)-Spiro[2-t-butyl-1,3-dioxolan-4-one-5,5′-4′,5′-dihydro-3′-methylisoxazole] 8
A solution of nitroethane (0.83 g, 11 mmol) and triethylamine (1 drop) in toluene (1 cm^3^) was stirred at room temperature, and a solution of phenyl isocyanate (1.3 g, 11 mmol) and 82 (1.6 g, 10 mmol) in toluene (1.2 cm^3^) was added dropwise over 1 h. An initial colour change from colourless to canary yellow along with rapid heating was observed, and a cold water bath was used to prevent boiling of the solvent. Once the reaction was deemed complete, the precipitate was filtered off and the solvent removed under vacuum. The resulting oil was taken up in hexane and CH_2_Cl_2_ and left to crystallise. The solid impurity was filtered off and the filtrate evaporated to give product 8 as an orange oil (1.46 g, 68%). [α]D +20.65 (c = 1.22, CH_2_Cl_2_). HRMS m/z (CI): found 214.1085. C_10_H_16_NO_4_ (M+H) requires 214.1079; ν_max_/cm^−1^ 1810 (C=O), 1763, 1720, 1299, 1233, 1164, 1072, 983 and 848; δ_H_ 0.97 (9H, s, t-Bu), 2.11 (3H, dd, J 1.1, 0.8, 3′-CH_3_), 3.07 (1H, half AB pattern of q, J 18.2, 0.8, 4′-CH_2_), 3.55 (1H, half AB pattern of q, J 18.2, 1.1, 4′-CH_2_) and 5.42 (1H, s, CH-t-Bu); δ_C_ 12.7 (3′-CH_3_), 23.1 (3CH_3_, t-Bu), 34.2 (C, CMe_3_), 45.5 (4′-CH_2_), 91.0 (5/5′-C), 108.8 (2-CH), 155.6 (C=N) and 167.2 (C=O); m/z (CI^+^) 214.11 (M+H^+^, 8%) and 128.03 (M^+^–Bu^t^CHO, 100%).
3.2.3. Preparation of (2S)-Spiro[2-t-butyl-1,3-dioxolan-4-one-5,3′-3′,4′-dihydro-5H-pyrazole] 9
The dioxolanone 2 (0.6 g, 3.8 mmol) was placed in a conical flask [important: no ground glass joints] with Et_2_O (10 cm^3^) in an ice bath, and an excess of diazomethane/Et_2_O [32] was distilled into the solution. Once this was complete, the reaction mixture was allowed to warm up to room temperature and stirred overnight to allow any unreacted gas to disperse. The solvent was then removed to yield the desired product 9 as a yellow viscous oil (0.62 g, 82%). HRMS m/z (CI): found 199.1092. C_9_H_15_N_2_O_3_ (M+H) requires 199.1083; ν_max_/cm^−1^ 1797 (C=O), 1721, 1366, 1289, 1223 and 1158; δ_H_ 1.03 (9H, s, t-Bu), 1.73 (1H, ddd, J 13, 9, 6, 4′-CH_2_), 2.20 (1H, ddd, J 13, 9, 4, 4′-CH_2_), 4.62 (1H, ddd, J 18, 9, 6, 3′-CH_2_), 4.87 (1H, ddd, J 18, 9, 4, 3′-CH_2_) and 5.84 (1H, s, CH-t-Bu); δ_C_ 23.1 (3CH_3_, t-Bu), 24.6 (4′-CH_2_), 34.7 (C, CMe_3_), 77.3 (3′-CH_2_), 110.5 (2-CH), 114.4 (5/5′-C) and 168.6 (C=O); and m/z (CI^+^) 199.11 (M+H^+^, 1%) and 95.02 (M^+^–CO, Bu^t^CHO, 100%).
3.2.4. Preparation of (2S)-Spiro[2-t-butyl-1,3-dioxolan-4-one-5,1′-2′,2′-diphenylcyclopropane] 10
The reactants 2 (0.9 g, 5.7 mmol) and diphenyldiazomethane (1.10 g, 5.7 mmol) were stirred in Et_2_O (10 cm^3^) for 5 days and monitored by TLC. No reaction had occurred, so the contents of the reaction vessel were transferred into a quartz tube and irradiated with a 400 W medium-pressure mercury lamp for 2 h. After this time, TLC analysis indicated that a reaction had taken place. The material was separated by flash column chromatography (SiO_2_, Et_2_O/hexane, 12:1) to furnish the product as a yellow/orange oil (0.25 g, 13%); HRMS m/z (CI): found 323.1642. C_21_H_23_O_3_ (M+H) requires 323.1647 ν_max_/cm^−1^ 3057, 1443, 1320 and 691; δ_H_ 0.96 (9H, s, t-Bu), 2.16 and 2.26 (2H, AB pattern, J 6.7, 3′-CH_2_), 5.38 (1H, s, CH-t-Bu), 7.27–7.35 (2H, m, Ph), 7.45–7.52 (3H, m, Ph), 7.56–7.63 (2H, m, Ph) and 7.78–7.84 (3H, m, Ph); δ_C_ 22.6 (3′-CH_2_), 23.2 (3CH_3_, t-Bu), 34.5 (C, CMe_3_), 44.1 (2′-C), 66.8 (5/1′-C), 109.2 (2-CH), 127.0 (CH, Ph), 127.4 (CH, Ph), 128.30 (2CH, Ph), 128.34 (CH, Ph), 128.7 (CH, Ph), 129.0 (CH, Ph), 129.2 (CH, Ph), 130.0 (2CH, Ph), 137.6 (C, Ph C-1), 138.8 (C, Ph C-1) and 173.0 (C=O); m/z (ES) 345.11 (M+Na^+^, 100%).
3.3. Pyrolysis of [3+2] Cycloadducts
3.3.1. Pyrolysis of (2S,5R)-Spiro[2-t-butyl-1,3-dioxolan-4-one-5,5′-4′,5′-dihydro-3′-phenylisoxazole] 7
FVP of the reactant 7 (0.1109 g, 0.40 mmol) was performed at 550 °C and 4.4 × 10^−1^ Torr. NMR spectroscopic analysis of the cold trap material indicated that no starting material remained. The substances present were pivalaldehyde (80%) [δ_H_ 1.08 (9H, s, t-Bu) and 9.48 (1H, s, CH=O); δ_C_ 23.3, 42.5 and 206.0] and benzonitrile (95%) [δ_H_ 7.46–7.50 (2H, m, Ph) and 7.60–7.69 (3H, m, Ph); δ_C_ 112.4 (C), 118.8 (C≡N), 129.0 (2CH), 132.1 (2CH) and 132.7 (CH)]
The formation of ketene was determined by repeating the pyrolysis and adding a small amount of ethanol directly into the cold trap immediately after isolation of the trap from the vacuum system. This gave ethyl acetate (40%) [δ_H_ 1.26 (3H, t, J 7.1, CH_3_), 2.04 (3H, s, CH_3_) and 4.12 (2H, q, J 7.1, CH_2_)].
3.3.2. Pyrolysis of (2S,5R)-Spiro[2-t-butyl-1,3-dioxolan-4-one-5,5′-4′,5′-dihydro-3′-methylisoxazole] 8
FVP of the reactant 8 (0.1454 g, 0.68 mmol) was performed at 550 °C and 4.4 × 10^−1^ Torr. ^1^H NMR spectroscopic analysis of the cold trap material indicated that no starting material remained. The substances present were pivalaldehyde (59%) [δ_H_ 1.08 (9H, s, t-Bu) and 9.48 (1H, s, CH=O)] and acetonitrile (54%) [δ_H_ 2.01 (3H, s, CH_3_)].
As described above, the formation of ketene was demonstrated by addition of a few drops of ethanol to the cold trap following pyrolysis. This gave ethyl acetate (23%) [δ_H_ 1.26 (3H, t, J 7.1, CH_3_) 2.04 (3H, s, CH_3_) and 4.12 (2H, q, J 7.1, CH_2_)].
3.3.3. Pyrolysis of (2S)-Spiro [2-t-butyl-1,3-dioxolan-4-one-5,3′-3′,4′-dihydro-5H-pyrazole] 9
FVP of the reactant 9 (0.0367 g, 0.185 mmol) was performed at 500 °C and 7.5 × 10^−1^ Torr. The product obtained was the spiro cyclopropane–dioxolanone 15 (ca. 40%) as an oil; ν_max_/cm^−1^ 1799 (C=O), 1286, 1151 and 1075; HRMS m/z (CI): found 171.1019. C_9_H_15_O_3_ (M+H) requires 171.1021; δ_H_ 0.99 (9H, s, t-Bu), 1.14–1.32 (4H, m, CH_2_-CH_2_) and 5.32 (1H, s, CH-Bu^t^); δ_C_ 11.3 (CH_2_), 13.2 (CH_2_), 23.3 (3CH_3_, t-Bu), 34.9 (C, CMe_3_), 60.0 (C-5) 109.4 (2-CH) and 174.3 (C=O); m/z (CI) 171 (M+H^+^, 100%). In addition, there were traces of 9 and pivalaldehyde [δ_H_ 1.08 (9H, s, t-Bu) and 9.48 (1H, s, CH=O)].
3.3.4. Pyrolysis of (2S)-Spiro[2-t-butyl-1,3-dioxolan-4-one-5,1′-2′,2′-diphenylcyclopropane] 10
FVP of the reactant 10 (0.08 g, 0.22 mmol) was performed at 500 °C and 4.3 × 10^−1^ Torr. NMR spectroscopic analysis of the cold trap material showed the presence of pivalaldehyde [δ_H_ 1.08 (9H, s, t-Bu) and 9.48 (1H, s, CH=O)] and several phenyl-containing materials. The separation of the non-volatile products by preparative TLC (Et_2_O/ hexane, 1:3) gave as the first fraction 4,4-diphenylbutenolide 18 (<5%) [δ_H_ 6.19 (1H, d, J 5.5, 2-CH), 7.94 (1H, d, J 5.5, 3-CH)]. The second, much larger fraction was a yellow oil (0.0365 g) which crystallised with time. This consisted, by NMR, mainly of benzophenone (75%) [δ_H_ 7.45–7.50 (4H, m, Ph), 7.56–7.62 (2H, m, Ph C-4) and 7.78–7.82 (4H, m, Ph); δ_C_ 128.3 (4CH, Ph), 130.0 (4CH, Ph), 1324 (2CH, Ph C-4), 137.6 (2C, Ph C-1) and 196.8 (C=O)], a little starting material, and a new product tentatively identified as 2-tert-butyl-5-(2,2-diphenylvinyl)-1,3-dioxolan-4-one 17 (7%) [δ_H_ 0.92 (9H, s, t-Bu), 5.12 (1H, d, J 9.9, 5-CH), 5.46 (1H, s, 2-CH), 6.12 (1H, d, J 9.9, 1′-=CH) and 7.18–7.35 (10H, m, Ph)].
3.4. X-Ray Structure Determination of Adduct 7
Data were collected on a Bruker SMART diffractometer using graphite-monochromated Mo Kα radiation, λ = 0.71073 Å. The data were deposited at the Cambridge Crystallographic Data Centre and can be obtained free of charge via http://www.ccdc.cam.ac.uk/structures. The structure was solved by direct methods and refined by full-matrix least squares against F^2^ (SHELXL, Version 2018/3 [35]).
(2S,5R)-Spiro[2-t-butyl-1,3-dioxolan-4-one-5,5′-4′,5′-dihydro-3′-phenylisoxazole] 7
Crystal data for C_15_H_17_NO_4_, M = 275.30, colourless prism, crystal dimensions 0.30 × 0.10 × 0.03 mm, orthorhombic, space group P2_1_2_1_2_1_ (No. 19), a = 5.7153(14), b = 11.497(3), c = 20.903(5) Å, V = 1373.5(6) Å^3^, Z = 4, Dc = 1.331 g cm^−3^, T = 93(2) K, R1 = 0.0513, Rw2 = 0.1029 for 2284 reflections with I > 2σ(I) and 182 variables, CCDC 2409957.
4. Conclusions
The first examples of [3+2] cycloaddition to the double bond of (2S)-5-methylene-2-t-butyl-1,3-dioxolan-4-one have been observed. The compound reacts well with nitrile oxides and diazo compounds but not with a range of other TAC types. NMR data have been documented for the novel spiro ring systems obtained, and the structure has been confirmed in one case by X-ray diffraction. Upon FVP, the adducts undergo a variety of processes involving interesting reactive intermediates whose reactivity has not been examined before. The addition of further types of TAC seems likely to provide an entry to more new chemistry of fused-ring heterocycles, and these results will be reported shortly.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Torres R.R. Spiro Compounds: Synthesis and Applications 1st ed.Wiley Hoboken, NJ, USA 2022444 p 10.1002/9781119567646 · doi ↗
- 2Ding A. Meazza M. Guo H. Yang J.W. Rios R. New development in the enantioselective synthesis of spiro compounds Chem. Soc. Rev.2018475946599610.1039/C 6CS 00825 A 29953153 · doi ↗ · pubmed ↗
- 3Zheng Y. Tice C.M. Singh S.B. The use of spirocyclic scaffolds in drug discovery Bioorg. Med. Chem. Lett.2014243673368210.1016/j.bmcl.2014.06.08125052427 · doi ↗ · pubmed ↗
- 4Seebach D. Naef R. Calderari G. α-Alkylation of α-heterosubstituted carboxylic acids without racemization: EPC-synthesis of tertiary alcohols and thiols Tetrahedron 1984401313132410.1016/S 0040-4020(01)82417-0 · doi ↗
- 5Calderari G. Seebach D. Asymmetrische Michael-Additionen. Stereoselektive Alkylierung chiraler, nicht racemischer Enolate durch Nitroolefine. Herstellung enantiomerenreiner γ-aminobuttersäure- und Bernsteinsäure-Derivate Helv. Chim. Acta 1985681592160410.1002/hlca.19850680611 · doi ↗
- 6Aitken R.A. Thomas A.W. Behaviour of dioxolanones as chiral acyl anion equivalents Synlett 1998199810210410.1055/s-1998-1569 · doi ↗
- 7Aitken R.A. Thomas A.W. Heterocyclic acyl and formyl anion equivalents Adv. Heterocycl. Chem.2001798911410.1016/S 0065-2725(01)79022-8 · doi ↗
- 8Zimmermann J. Seebach D. Brominations of cyclic acetals from α-amino acids and α- or β-hydroxy acids with N-bromosuccinimide Helv. Chim. Acta 1987701104111410.1002/hlca.19870700423 · doi ↗