Breaking Grounds: A Comprehensive Analysis of Cutting-Edge Treatments for Primary Biliary Cirrhosis/Primary Biliary Cholangitis With Futuristic Treatments
Asad Ali Khan, Furqan Ul Haq, Qazi Muhammad Farooq Wahab, Taimur Aslam, Azeem Khalid, Asad Ali

TL;DR
This paper reviews current and emerging treatments for primary biliary cholangitis, emphasizing personalized strategies and future therapeutic options.
Contribution
The paper provides a comprehensive overview of new diagnostic approaches and treatment advancements in PBC management.
Findings
Elafibranor is FDA-approved and effective based on the ELATIVE trial.
Seladelpar is under FDA review and shows promise in the ENHANCE III trial.
Immunotherapies like rituximab and budesonide lack clinical significance in PBC treatment.
Abstract
Primary biliary cholangitis (PBC) is an autoimmune disorder characterized by biliary destruction leading to intrahepatic biliary cholestasis. It predominantly affects women during the fifth and sixth decades. Treatment options have progressed from ursodeoxycholic acid (UDCA) and obeticholic acid (OCA) to liver and stem cell transplant. The objectives include summarizing established and new diagnostic approaches for PBC along with reviewing efficacy treatments, their side effects, and future directions. The treatment of PBC is based on risk stratification, including assessment of the patient’s age, sex, clinical pattern, biochemical and antibody profile, histology, and markers of fibrosis. UDCA and OCA are Food and Drug Administration (FDA) approved first-line and second-line agents. Elafibranor, a recently FDA-approved agent based on its efficacy, was shown in the ELATIVE trial.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
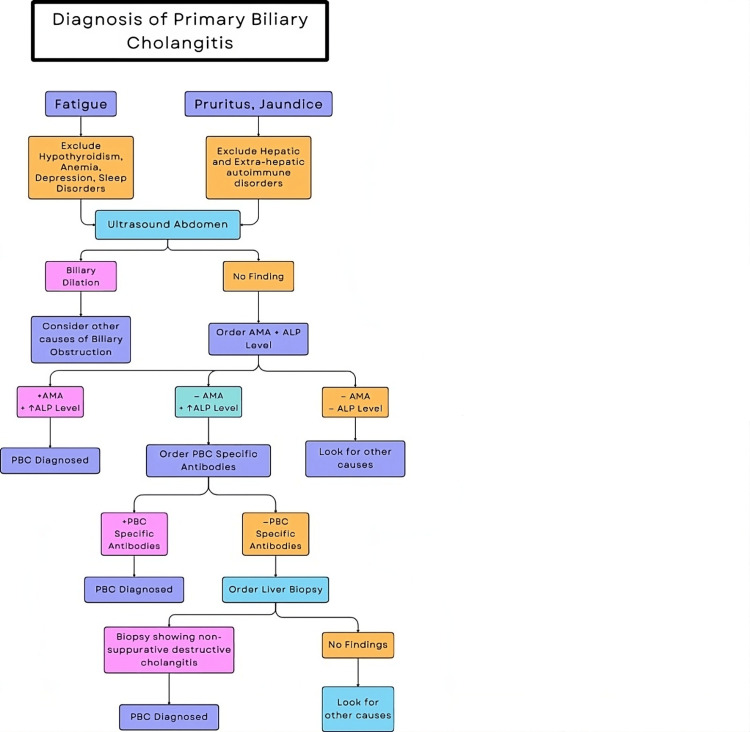 Figure 1
Figure 1| Serial no. | Factors | Low risk | High risk |
| 1 | Age | ˃55 years | ˂55 years |
| 2 | Sex | Female | Male |
| 3 | Clinical pattern | No symptoms | Symptomatic disease |
| AIH/PBC OS | |||
| Premature ductopenic varient | |||
| 4 | Antibody profile | AMA | Anti-gp210+ |
| ACA+ | |||
| Anti-HK1+ | |||
| Anti-KLHL12+ | |||
| 5 | Biochemical panel | Normal bilirubin | ↑ Bilirubin |
| ALP ˂ 2× ULN | ALP ≥ 2× ULN | ||
| APRI score ˃ 0.54 | |||
| 6 | Histology | No/mild fibrosis | Advanced fibrosis/cirrhosis |
| Interface hepatitis | |||
| Ductopenia at the diagnosis | |||
| 7 | Non-invasive markers of fibrosis | LSM < 8 kPa/↑ < 2.1 kPa/y | LSM > 15 kPa/↑ > 2.1 kPa/y |
| ELF score < 10.0 | ELF score ≥ 10.0 | ||
| MRE < 4.6 kPa | MRE > 4.6 kPa |
| Characteristics | Drug | Mechanism | Effects | Clinical status | Dose | Adverse effects |
| FDA-approved drugs | Ursodeoxycholic acid (approved in 1997) | Acts in the liver through multiple interrelated pathways | Promotes bile acid excretion, decreases biliary cholesterol, reduces liver damage by bile acid, and improves overall and LTF survival | FDA-approved first-line agent | 13-15 mg/kg/day | Nausea, vomiting, diarrhea, flatulence, and sleep problems |
| Obeticholic acid (approved in 2016) | Farnesoid X receptor agonist | Inhibits bile acid synthesis, improves enterohepatic circulation, and has anti-inflammatory and anti-fibrotic effects | FDA-approved second-line agent | 5-10 mg/day | Pruritus, hepatic decompensation, and lipid derangements | |
| Elafibranor (approved in 2024) | PPARα and δ agonist | Reduction in the ALP, triglycerides, and cholesterol levels | Recently approved by the FDA based on the ELATIVE trial results | 80 mg/day | Headache, nausea, fatigue, diarrhea, and creatinine elevation | |
| Potential future therapies | Seladelpar (MBX-8025) | PPARδ agonist | Improvement in ALP and pruritis | Under FDA review for approval in the ENHANCE phase III trial | 10 mg/day | Nil reported |
| Bezafibrate | pan-PPAR agonist | Improvement in liver biochemistry, fibrosis, and stiffness and alleviates pruritus | Third-line treatment in the BEZURSO trial adds on therapy with UCA | Trial with 400 mg/day | Myalgia, increased creatinine, and hepatotoxicity | |
| Fenofibrate | PPARα agonist | Improves liver biochemistry, pruritus, and LTF survival and reduces fibrosis | ChiCTR1800020160 trial | Tested dose is 200 mg/day | NA | |
| Pemafibrate | PPARα agonist | Improvement in ALP and GGT | NA | NA | ||
| Tropifexor (LJN452) | Farnesoid X receptor agonist | Prevents bile acid-mediated liver damage and fibrosis | Under clinical trials and | 30-90 ug daily doses have been tested | Pruritus | |
| Cilofexor (GS-9674) | Farnesoid X receptor agonist | Reduction in ALP levels observed | Phase II clinical trial ( | 30 and 100 mg doses have been tested | Pruritis | |
| Aldafermin (NGM282) | FGF19 analogue | Improvement in ALP, transaminase, and liver damage and protection from fibrosis and HCC progression | 0.3 and 3 mg dose groups | NA | ||
| Saroglitazar | PPARα and γ agonists | Improvement in ALP and cholestatic markers | 1 and 2 mg doses are being tested | Elevation in aminotransferases | ||
| Linerixibat (GSK2330672) | ASBT inhibitor | Reduces bile acid and improves pruritus | GLISTEN is an ongoing phase III trial ( | NA | NA | |
| Volixibat | ASBT inhibitor | - | NA | NA | ||
| Setanaxib (GKT137831) | Inhibits NOX1 and 4 | Improves cholestasis, liver fibrosis, and fatigue | Doses of 400-1600 mg have been tested | NA | ||
| Ineffective therapies | Rituximab | Anti-CD20 mab | Improves ALP levels but no clinically significant effects seen | 1000 mg tested | NA | |
| Budesonide | Immunosuppressant | Improvement in cholestatic markers | Tested dose is 9 mg/day | Arthralgia, hypertension, muscle spasms, weight gain, peripheral edema, and dyspepsia | ||
| Preclinical agents | BAR501INT-777 and INT-767 | TGR5 agonists | Reduces inflammation | - | - | - |
| 5-Aza-2-deoxycytidine (DAC) | Maintains balance of Treg/Th17 | Anti-inflammatory and promotes tolerance | - | - | - | |
| CNP-104 nanoparticle | Targets PDC-E2 | - | - | - | - | |
| Simtuzumab | Monoclonal antibody against LOXL2 | Improves liver fibrosis | - | - | - |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLiver Diseases and Immunity · Liver Disease Diagnosis and Treatment · Pediatric Hepatobiliary Diseases and Treatments
Introduction and background
Primary biliary cholangitis (PBC), previously called primary biliary cirrhosis, is a rare autoimmune chronic hepatobiliary disorder in which there is progressive inflammatory biliary epithelial cell (BEC) destruction, cholestasis, cirrhosis, and ultimately liver failure [1]. Although it can affect anyone of any age or gender, the condition is more prevalent in women and those over 50 years. It affects people of all races, ethnicities, and countries worldwide, and its incidence and prevalence are rising, particularly among women [2,3].
Both genetic and environmental factors are responsible for the pathogenesis of PBC. In PBC, a disease-specific autoantibody called anti-mitochondrial antibody (AMA) targets the cellular mitochondria membranes. This anti-mitochondrial response is primarily responsible for disease pathogenesis, although some non-autoimmune mechanisms may also play a role [4]. Smoking, infections, hormonal therapy, and nail polishing are usually the environmental culprits for the disease pathophysiology. More than 60% of patients are asymptomatic and are diagnosed accidentally when they are found to have abnormal labs, i.e., elevated alkaline phosphatase (ALP), positive AMA, or cholestatic liver pattern labs, by chance [5]. Those who are typically symptomatic manifest with signs and symptoms of chronic cholestasis, i.e., fatigue, pruritus, jaundice, xanthomas, xanthelasmas, and asthenia. Diagnosis of PBC is established with a detailed history, physical examination, lab tests [anti-nuclear antibody (ANA), AMA, ALP, and gamma-glutamyl (GGT)], imaging tests [magnetic resonance cholangiopancreatography (MRCP) and ultrasound (US)], and liver biopsy [6].
A liver biopsy is not required for PBC diagnosis, and diagnosis can be established without a liver biopsy if other criteria are met. Also, AMA positivity alone is not sufficient to make the diagnosis of PBC. Complications of PBC include hepatocellular complications and systemic complications [7]. Hepatocellular complications include cirrhosis, portal hypertension, and hepatocellular carcinoma. Systemic complications include cognitive dysfunction, sicca syndrome, arrhythmia and ventricular dysfunction, nephropathy, osteoporosis, fractures, and increased risk of malignancies [8].
Review
Methods
This review concentrates on treatment options and breakthroughs in the diagnosis of PBC. Using precise keywords and MeSH phrases linked to PBC diagnosis and therapies, a thorough search was conducted throughout PubMed, Cochrane Library, EMBASE, and ClinicalTrials.gov. Cohort studies, phase II/III clinical trials, and randomized controlled trials were among the studies that qualified for inclusion, while case reports and animal studies were excluded. Study design, patient demographics, diagnostic techniques, treatments (first-line, second-line, and novel agents), and clinical outcomes were all covered by the data extraction process.
Epidemiology
The disease is more common among females, with a female-to-male ratio ranging from 4 to 5:1 and 9 to 10:1, and those above 50 years, but it can occur across any age and gender group [9]. The incidence and prevalence of PBC are on the rise, especially among the female patient population. The average incidence rate is 3.0 cases per 100,000 individuals per year, with a prevalence rate of 21.05 cases per 100,000 individuals. PBC affects individuals of all ethnicities, races, nationalities, and distribution worldwide [10].
Etiology and pathophysiology of PBC
The exact etiology and mechanism by which PBC develops remain unclear. PBC is a multifactorial disease, and both genetic and environmental factors are involved in the disease pathophysiology. In PBC, the primary target of damage is to the BECs. This has been associated with the immunobiology of BECs in persons who are genetically predisposed and subjected to environmental stressors. BECs overexpress microRNA-506 (miR-506), resulting in inadequate secretion of biliary bicarbonate secretion that manifests as cholestasis. In addition, it targets exclusively the messenger ribonucleic acid (mRNA) of anion exchanger 2 (AE2), resulting in cholangiocytes developing PBC-like characteristics. The E2 component of the pyruvate dehydrogenase complex (PDC-E2) is overexpressed and exhibits odd localization as a result of toxic bile acid’s (BA) unregulated infiltration of BECs and promotion of apoptosis. Due to the absence of glutathionylation in apoptotic bodies, PDC-E2 is unaltered during cholangiocyte apoptosis. Circulating AMA would identify the PDC-E2 found in the apoptotic bodies, and the immune complex would then activate the innate immune system in an individual with a genetic predisposition.
Clinical presentation and diagnosis of PBC
Most of the patients with PBC are asymptomatic at the time of diagnosis. Symptomatic patients present with pruritic and fatigue, which are present in up to 70% of patients. Thyroid disorders, anemia, depression, adrenal disorders, and sleep disorders could also cause fatigue and should be considered before attributing symptoms to PBC [4]. Sjogren syndrome, thyroid disorders, celiac disease, and systemic sclerosis are the common autoimmune disorders commonly seen in patients with PBC [11]. In all, 90 to 95% of patients with PBC have positive serology for AMA. Because of its high specificity, PBC diagnosis can be easily made in a patient presented with positive AMA serology and cholestasis without ordering a liver biopsy [12]. Figure 1 presents the algorithmic approach for the diagnosis.
Algorithm for the diagnosis of PBCALP, alkaline phosphatase; PBC, primary biliary cholangitis; AMA, anti-mitochondrial antibody
PBC-specific anti-nuclear antibodies like anti-sp100 and anti-gp210 can be found in 50% of AMA-negative patients. This helps in the diagnosing of PBC in AMA-negative individuals [13]. Anti-kelch-like 12 (KLHL12) and anti-hexokinase 1 (HK1) are other PBC-specific antibodies that can be found in PBC patients, but their practical use needs further studies and understanding. The nature of disease and clinical outcomes is exhibited similarly by both AMA-positive and AMA-negative PBC patients [14]. The risk of fat-soluble vitamin deficiencies and lipid disorders is high in PBC patients. Unless there is the absence of serological markers or high suspicion of nonalcoholic steatohepatitis or autoimmune hepatitis, a liver biopsy is not needed for the diagnosis of PBC [15]. The diagnosis of PBC is supported by the absence of other liver and systemic pathologies and chronically high ALP levels but positive serology for AMA. It is also supported by the absence of AMA but positive PBC-specific antibodies in patients with chronically high ALP levels and liver biopsy showing evidence of PBC in a patient with elevated ALP levels without any serological markers H.
Risk stratification and survival analysis
The progression of PBC varies throughout the population; while some see a gradual decline in their condition, others experience severe fibrosis and liver cirrhosis in a matter of years. To determine a patient’s prognosis, it is necessary to evaluate their unique risk of progression. In order to do this, a number of laboratory, serological, clinical, and demographic factors are assessed. Additionally, the disease stage is determined depending on the degree of fibrosis and the patient’s reaction to treatment. Table 1 summarizes the risk stratification [16,17].
Hardie et al. identified PBC patients as low-risk if they have a positive outcome in terms of improvement and as high-risk if they have a poor outcome in terms of PBC development [18]. It is reported that genes associated with apoptosis and cell cytotoxicity are up-regulated, while genes related to complement pathways are down-regulated in high-risk disease patients as compared to low-risk patients. In high-risk patients, the pathways related to T-cell activation, leukocyte migration, apoptosis, and interferon-gamma response were significant. The gene product of CDKN1a, the senescence marker p21WAD1/Cip, shows higher expression levels on the bile ducts in high compared to low-risk patients [18].
Tian et al. carried out a study to explore novel biomarkers of risk stratification for PBC patients utilizing the gene expression omnibus (GEO) database. They identified 166 differentially expressed genes (DEGs), and 15 of them were associated with disease progression (seven up-regulated genes and eight down-regulated genes). Four core-risk-related genes, including TXNIP, CD44, ENTPD1, and PDGFRB, were defined, and three of these genes (TXNIP, CD44, and ENTPD1) showed upregulation on qRT-PCR resulting in the development of a three-gene panel for screening high-risk PBC patients [19].
Treatment of PBC
The treatment of PBC is based on risk stratification, which includes assessment of the patient’s age, sex, clinical pattern, biochemical profile, histology, antibody profile, and markers of fibrosis; based on these, the patients are divided into low-risk and high-risk for PBC [20].
For the past few decades, different regimes have been used for PBC. Ursodeoxycholic acid (UDCA) is a Food and Drug Administration (FDA) approved first-line agent acting on the promotion of BA excretion and decreases cholesterol, which reduces biliary damage with a dose of 13-15 mg/kg/day, slightly risk of nausea and vomiting though flatulence and diarrhea are common [20]. Obeticholic acid (OCA) is an FDA-approved second-line agent with a dose of 5-10 mg/day, though it causes pruritus and lipid derangement. The efficacy of Elafibranor, a recently FDA-approved agent, is shown in ELATIVE trial results with a dose of 80 mg/day, which causes headache, diarrhea, and raised creatinine. Seladelpar, which is under review for FDA approval in the ENHANCE III trial, is also used in PBC at a dose of 10 mg/day. Fibrate, a third-line treatment like bezafibrate, fenofibrate, or pemafibrate, was tried and found efficacious in different trials, such as the BEZURSO trial, ChiCTR1800020160 trial, and NCT06247735 that is an ongoing phase II trial.
Tropifexor is in phase II clinical trial, is under review, and is used in a daily dose of 30-90 ug, which causes pruritus [21]. Cilofexor is also in phase II clinical trial and is used in a dose of 30-100 mg, which also causes pruritus. Linerixibat, an apical sodium-dependent BA transporter (ASBT) inhibitor, and volixibat, an ASBT inhibitor, are under phase III and II trials, respectively. Setanaxib, which is a NOX 1 and 4 inhibitor, is also under phase II trial [22].
The question of whether we use immunotherapy has been answered in the NCT02376335 and NCT00746486 trials, stating that we cannot use rituximab and budesonide as no clinical significance is seen, along with under power study. Other immunosuppressive and monoclonal antibodies, such as BAR501INT-777 and INT-767, 5-aza-2-deoxycytidine, and CNP-104 nanoparticles, are under study in the treatment of PBC, and their results are awaited [23].
Discussions
PBC is an autoimmune disease, though the treatment is based on improving liver functions and slowing down the progression of worsening liver function tests. The treatment landscape has significantly evolved over the recent decades, reflecting advancement in our understanding of the disease pathophysiology and patient needs. This review synthesizes the current evidence on therapeutic options, highlighting both the established and emerging treatments [24].
Most PBC patients respond well to UDCA, as one study has shown improved survival without transplantation in patients treated with UDCA compared to untreated patients [hazard ratio (HR): 0.46; 95%CI, 0.40 to 0.52; p < 0.001). However, some patients develop advanced liver cirrhosis even after receiving treatment. Multiple studies have confirmed its efficacy in improving biochemical markers, slowing disease progression, and enhancing survival rates [25]. The dosage is kept between 13 and 15 mg/kg and can be given as a single dose orally. The majority of response criteria take into account both bilirubin and ALP, which are useful in clinical practice (higher ALP readings may be utilized as a risk marker of early but quickly progressing PBC, whereas bilirubin tends to be an indicator of advanced disease). UDCA is the first-line approved drug, though it causes nausea, vomiting, flatulence, and diarrhea. The need for second-line treatment has been recommended after the use of UDCA [22].
OCA has emerged as the first approved second-line therapy in patients with inadequate response to UDCA. Clinical trials have demonstrated its efficacy in further reducing ALP levels and other markers of cholestasis. OCA acts by activating the farnesoid X receptor (FXR), which regulates BA synthesis and transport. However, concerns about pruritus and potential long-term cardiovascular risks necessitate careful patient selection and monitoring. This review highlights the importance of personalized therapy, considering both the benefits and potential side effects of OCA [26].
Fibrates, particularly fenofibrate and bezafibrate, have promise in PBC treatment, especially in combination with UDCA. These agents, which activate peroxisome proliferator-activated receptors (PPARs), have demonstrated improvements in liver biochemistry and symptoms. However, their impact on long-term outcomes, such as liver transplantation rates and overall survival, requires further investigation. Ongoing studies and real-world evidence are essential to establish their role in the standard treatment algorithm [27].
In emerging therapies, there are several novel therapeutic agents currently under investigation, targeting various pathways involved in PBC pathogenesis. Among these, seladelpar, a potent PPAR-delta agonist, has shown efficacy in reducing ALP, marked improvement in liver biochemistry, and improved liver histology with a favorable safety profile [28]. Another novel agent is norUDCA, which is a modified BA with anti-inflammatory and anti-fibrotic properties and is currently in clinical trials with the dose maintained at 1500 mg, resulting in a significant reduction of ALT within 12 weeks of treatment as compared to placebo.
Liver-Targeted Immunotherapies
Liver-targeted immunotherapies are designed to modulate the immune response specific to PBC, offering the potential for disease modification. The review discusses the potential of these emerging therapies to address unmet needs in PBC treatment, particularly for patients who do not respond adequately to existing therapies. Clinical trials of pipeline medications and animal research have shown encouraging results in halting the course of illness. In the early stage of the disease, treatment is targeted toward immune-mediated pathogenesis and anti-inflammatory therapies. On the other hand, in the late stage of the disease, defined by fibrosis and cirrhosis, anti-cholestatic and anti-fibrotic therapies are utilized [29].
In combination therapy for PBC, given the heterogeneity in treatment response, combination therapy represents a promising approach. Combining agents with complementary mechanisms of action could enhance therapeutic efficacy and mitigate adverse effects. Preliminary studies on combinations, such as UDCA with fibrates or OCA, have shown encouraging results, but larger, long-term studies are needed to confirm these findings [30].
Future directions
Future research should focus on understanding the predictors of treatment response, optimizing combination therapies, and developing biomarkers for early diagnosis and treatment monitoring. Moreover, the integration of patient-reported outcomes in clinical trials will provide insights into the impact of therapies on quality of life. Collaborative efforts in multicenter trials and real-world studies are essential to advance the treatment paradigm for PBC.
Conclusions
This review highlights the significant progress made in the treatment of PBC while acknowledging the challenges that remain. The emergence of new therapies and the potential of combination treatments offer hope for improving outcomes for all patients with PBC. Personalized treatment strategies, continuous monitoring, and a comprehensive approach to symptom management are key to optimizing care and enhancing the quality of life for individuals affected by this chronic liver disease.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Definition and management of patients with primary biliary cholangitis and an incomplete response to therapy Clin Gastroenterol Hepatol Montano-Loza AJ Corpechot C 224122511920213262912510.1016/j.cgh.2020.06.062 · doi ↗ · pubmed ↗
- 2The British Society of Gastroenterology/UK-PBC primary biliary cholangitis treatment and management guidelines Gut Hirschfield GM Dyson JK Alexander GJ 156815946720182959306010.1136/gutjnl-2017-315259 PMC 6109281 · doi ↗ · pubmed ↗
- 3Worldwide incidence of autoimmune liver disease Dig Dis Jepsen P Grønbæk L Vilstrup H 21233 Suppl 2201510.1159/00044070526641102 · doi ↗ · pubmed ↗
- 4Primary biliary cholangitis: 2018 practice guidance from the American Association for the Study of Liver Diseases Hepatology Lindor KD Bowlus CL Boyer J Levy C Mayo M 3944196920193007037510.1002/hep.30145 · doi ↗ · pubmed ↗
- 5Diagnosis and management of primary biliary cholangitis Am J Gastroenterol Younossi ZM Bernstein D Shiffman ML Kwo P Kim WR Kowdley KV Jacobson IM 486311420193042959010.1038/s 41395-018-0390-3 · doi ↗ · pubmed ↗
- 6Systemic complications of primary biliary cholangitis Clin Liver Dis Zapata M Pagan-Torres H Mayo MJ 1151282820243794515310.1016/j.cld.2023.07.004 · doi ↗ · pubmed ↗
- 7Regarding: risk of fractures and postfracture mortality in 3980 people with primary biliary cholangitis J Intern Med Chang HC Gau SY 53629420233727645510.1111/joim.13676 · doi ↗ · pubmed ↗
- 8Managing the symptoms and complications of cholestasis Clin Liver Dis (Hoboken) Pedersen MR Mayo MJ 1201241520203225712310.1002/cld.901PMC 7128033 · doi ↗ · pubmed ↗