In Vitro Activity of the Triazinyl Diazepine Compound FTSD2 Against Drug-Resistant Mycobacterium tuberculosis Strains
Carlos Aranaga, Ruben Varela, Aura Falco, Janny Villa, Leydi M. Moreno, Manuel Causse, Luis Martínez-Martínez

TL;DR
A new compound called FTSD2 shows strong activity against drug-resistant tuberculosis bacteria without harming human cells.
Contribution
FTSD2's selective antitubercular activity against drug-resistant strains and low cytotoxicity is newly demonstrated.
Findings
FTSD2 inhibited drug-resistant M. tuberculosis growth at low concentrations (0.5-1 mg/L).
FTSD2 showed concentration-dependent bactericidal activity and no cytotoxicity at concentrations below 64 mg/L.
FTSD2 suppressed intracellular M. tuberculosis growth in macrophages after 192 hours.
Abstract
Background/Objectives: Compounds derived from pyrimido-diazepine have shown selective inhibition of the susceptible Mycobacterium tuberculosis strain H37Rv. However, there is a need for studies that evaluate the activity of these compounds against multidrug-resistant strains and clinical isolates. This study aims to evaluate the antitubercular potential of FTSD2 against drug-resistant strains of M. tuberculosis. Methods: The compound 4-(2,4-diamino-8-(4-methoxyphenyl)-8,9-dihydro-7H-pyrimido[4,5-b][1,4]diazepin-6-yl)-N-(2-(4-(dimethylamino)-6-(4-fluorophenyl)amino-1,3,5-triazin-2-yl)amino)ethyl)benzenesulfonamide (FTSD2) was tested against drug-resistant M. tuberculosis strains at minimal inhibitory and bactericidal concentrations (MIC and MBC). Kill curve assays were performed to assess bactericidal activity, and cytotoxicity was evaluated in human monocyte-derived macrophages and the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
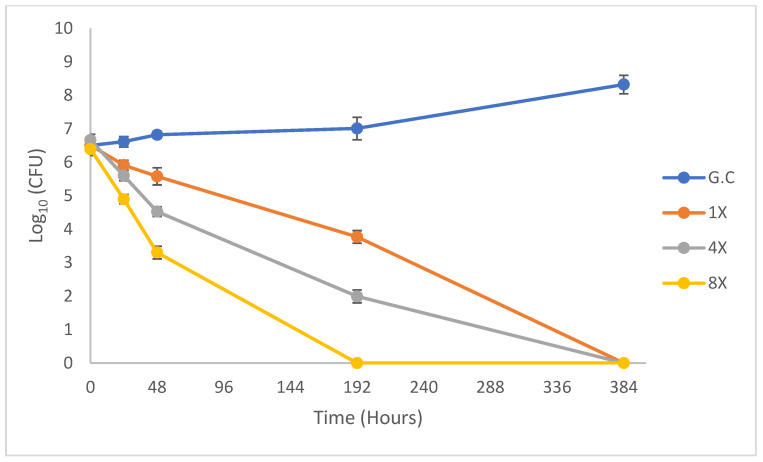 Figure 1
Figure 1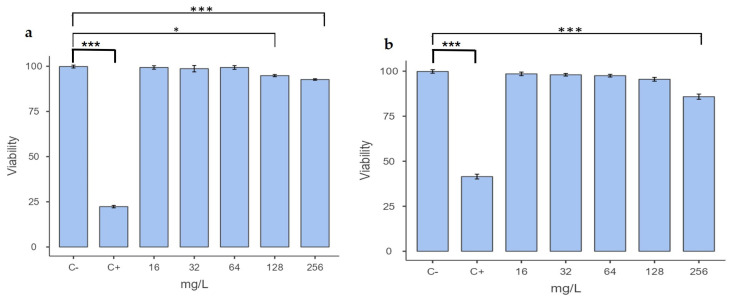 Figure 2
Figure 2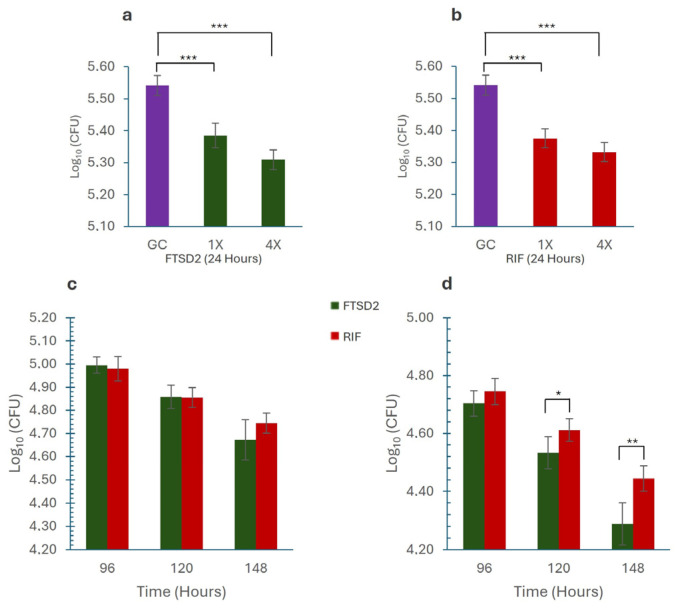 Figure 3
Figure 3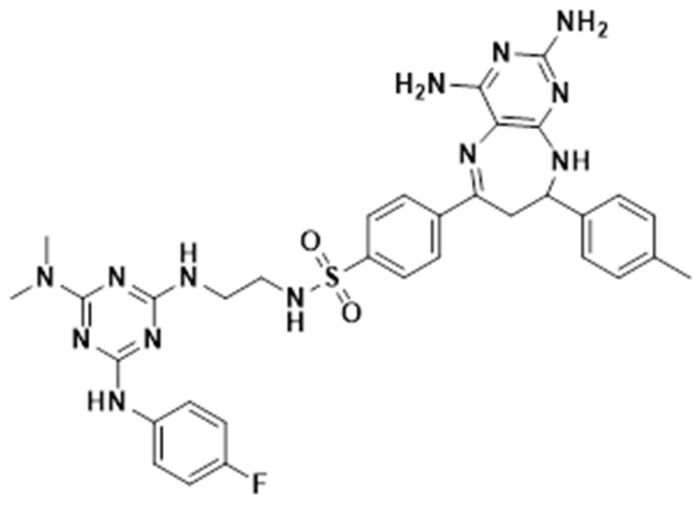 Figure 4
Figure 4- —Dirección General de Investigaciones of Universidad Santiago de Cali
- —research Group GC24 “Clinical and Molecular Microbiology” of the Instituto Maimónides de Investigación Biomédica (IMIBIC)
- —Asociación de Universitaria Iberoamericana de Posgrados
- —Corporación Universitaria Remington
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSynthesis and Characterization of Heterocyclic Compounds · Tuberculosis Research and Epidemiology · Quinazolinone synthesis and applications
1. Introduction
With significant advances in the diagnosis and treatment of tuberculosis (TB) in recent years, new options have been developed to combat the disease. However, it remains one of the leading causes of death worldwide due to a single infectious agent [1]. The introduction of new drugs, such as bedaquiline [2], delamanid [3], and pretomanid [4], as well as the repositioning of existing drugs, such as fluoroquinolones, linezolid, clofazimine, and streptomycin, have provided new options for improving the treatment of drug-resistant tuberculosis. In response to this situation, the World Health Organization (WHO) has updated treatment regimens for patients with drug-resistant tuberculosis, emphasizing the use of levofloxacin in cases of isoniazid-monoresistant tuberculosis, as well as the combination of levofloxacin or moxifloxacin, bedaquiline, and linezolid in patients with rifampicin-resistant or multidrug-resistant (MDR) tuberculosis [5]. Despite these advances, significant challenges remain. In 2023, the World Health Organization reported over 400,000 cases of drug-resistant tuberculosis, emphasizing the urgent need for novel therapeutic options to address this growing challenge [6].
Bedaquiline is one of the most widely used new bactericidal drugs for treating patients with MDR-TB [7]. It binds to ATP synthase in both replicating and latent mycobacteria [8]. However, mutations in the atpE gene, which encodes the C subunit of mycobacterial ATP synthase, have been shown to confer resistance to this drug [9]. Other mutations, such as those associated with the pepQ and mmpR genes, may be involved in a moderate increase in the minimum inhibitory concentration (MIC) of both bedaquiline and clofazimine [9,10]. Similarly, mutations associated with the fbiA, fbiC, ddn, and fgd1 genes are responsible for resistance to delamanid and pretomanid [9], which are alternatives for patients who cannot be treated solely with drugs from group A (Levofloxacin, moxifloxacin, Bedaquiline, and linezolid) and group B (Clofazimine, Cycloserine, and terizidone) in a prolonged treatment regimen. The emerging resistance to these drugs, as well as to repurposed drugs [7,11,12,13,14], and the toxicity of current TB treatment regimens for some patients continue to limit therapeutic options and present significant challenges in TB care [6]. These findings highlight the need to continue searching for new compounds that can cure all patients.
In a recent study, a series of triazinylamino chalcones exhibited activity by inhibiting the in vitro growth of M. tuberculosis H37Rv, with minimum inhibitory concentrations (MICs) ranging from 25 to 50 mg/L. Based on these structures, a series of seven triazinylamino-pyrimido[4,5-b][1,4]diazepines was synthesized, showing a 4- to 10-fold reduction in MIC compared to that of the initial triazinylamino chalcones. Among these compounds, FTSD2 (4-(2,4-diamino-8-(4-methoxyphenyl)-8,9-dihydro-7H-pyrimido[4,5-b][1,4]diazepin-6-yl)-N-(2-(4-(dimethylamino)-6-(4-fluorophenyl)amino-1,3,5-triazin-2-yl)amino)ethyl)benzenesulfonamide) was the most active, with MIC values 2 to 4.5 times lower than the other compounds in the series [15].
Additionally, FTSD2 showed no activity against bacteria such as Staphylococcus aureus, Pseudomonas aeruginosa, Klebsiella pneumoniae, and Escherichia coli, even at concentrations exceeding 100 mg/L. This selectivity is particularly relevant in the development of new drugs, as antitubercular treatments are typically prolonged and can affect the patient’s normal microbiota [15].
In contrast, various compounds with heterocyclic systems, including triazines or pyrimido[4,5-b][1,4]diazepine structures, have demonstrated anticancer activity, partly due to their ability to inhibit serine/threonine kinases [16,17,18]. Mycobacterium tuberculosis possesses two essential serine/threonine kinases for in vitro survival [19], whereas many other bacteria rely on phosphorylation systems mediated by histidine kinases [20]. Although the molecular target of FTSD2 has not been determined, its selective activity against M. tuberculosis suggests that it may act through a mechanism different from that of conventional drugs. In this regard, further characterization of its activity will be key to a better understanding of its therapeutic potential.
However, the standard treatment regimen for drug-susceptible tuberculosis has a 99% efficacy rate. Therefore, efforts to control tuberculosis have focused on finding alternative treatments for rifampicin-resistant tuberculosis (RR-TB), multidrug-resistant tuberculosis (MDR-TB), and extensively drug-resistant tuberculosis (XDR-TB). In this study, we determined the in vitro activity of the triazinyl diazepine compound FTSD2 against drug-resistant reference strains of M. tuberculosis and clinical isolates to assess its potential as an antitubercular drug [15,21].
2. Results
2.1. Susceptibility Testing
The MIC of FTSD2 for M. tuberculosis, M. avium, and rapidly growing mycobacteria (RGM) is shown in Table 1. For M. tuberculosis strains and clinical isolates, the MIC ranged from 0.25 to 1 mg/L. In contrast, the MIC for clinical isolates of M. avium and RGM was higher than 100 mg/L, which was the maximum concentration tested in the assays. The MBC/MIC ratio for M. tuberculosis was 1, indicating that FTSD2 acts as a bactericidal antibiotic.
2.2. Dose−Response of M. tuberculosis
The time-kill (TK) kinetics of FTSD2 against M. tuberculosis H37Rv are presented in Figure 1. The results showed notable bactericidal activity depending on the concentration from 24 h, with a decrease in bacterial concentration of 0.5, 1.1, and 1.5 logarithmic units (Log10) for concentrations of 1× MIC, 4× MIC, and 8× MIC, respectively (Supplementary Material Table S1: Time-kill). At 196 h, no bacterial growth was observed, indicating a reduction of 6.3 Log_10_, while for the same period, the reduction in growth at concentrations of 1× MIC and 4× MIC was 2.9 and 4.7 Log_10_. After 384 h, no bacterial growth was observed at any of the concentrations (Figure 1).
2.3. Determination of Cytotoxicity
Cell viability assays were performed on human monocyte-derived macrophages (hMDM) and the murine RAW 264.7 macrophage cell line (Figure 2). No significant differences in viability were observed between the growth control and 16, 32, and 64 mg/L concentrations in either cell type. Greater sensitivity to FTSD2 was observed in hMDM, with a statistically significant difference (p = 0.015) between the 128 mg/L concentration and the growth control (Figure 2a), while in RAW 264.7 macrophages, significant differences (p < 0.001) were only observed at 256 mg/L compared to the growth control (Figure 2b. Supplementary Material Table S2). These results highlight the compound’s high therapeutic index, which combines potent antimycobacterial activity with minimal cytotoxicity.
2.4. Macrophage Assay
The bactericidal activity of FTSD2 was evaluated using rifampicin as a control against M. tuberculosis H37Rv in a macrophage infection model, employing concentrations of 1× MIC (1 mg/L FTSD2, 0.2 mg/L RIF) and 4× MIC (4 mg/L FTSD2, 0.8 mg/L RIF). At 24 h, both concentrations significantly reduced bacterial growth compared to that in the control (p < 0.001) (Figure 3a,b). This trend persisted throughout the experiment, with more pronounced inhibition observed at 4× than at 1× at all time points, as evidenced by the TK assays. At 1× concentration, FTSD2 exhibited inhibition comparable to that of rifampicin, with no significant differences. However, at 4× concentration, FTSD2 demonstrated greater inhibition, with statistically significant differences at 120 h (p = 0.018) and 148 h (p = 0.001) compared to rifampicin (Figure 3c,d. Supplementary Material Table S3), suggesting that FTSD2 may sustain its activity for a longer period compared to rifampicin, an aspect of potential relevance for its further development.
3. Discussion
The selection of drug-resistant strains currently used against M. tuberculosis remains one of the biggest challenges in controlling TB. In 2022 alone, 410,000 cases of TB caused by drug-resistant strains were reported, of which 175,650 patients began treatment [6]. To achieve therapeutic success in managing this resistance, it is essential to have a more significant number of antimicrobials that increase efficacy and reduce toxicity in patients [22]. In this context, we evaluated the potential of a triazinyloxy-pyrimido[4,5-b][1,4]diazepine compound (FTSD2) to inhibit the growth of drug-resistant M. tuberculosis strains, including clinical isolates.
The results showed that the minimum inhibitory concentration (MIC) for resistant strains was similar to that observed for susceptible strains H37Ra and H37Rv, with values ranging from 0.25 to 1 mg/L (Table 1). These MICs are comparable to those of several clinically used drugs, such as the aminoglycoside amikacin (1 mg/L), fluoroquinolones ciprofloxacin, ofloxacin, and levofloxacin (0.5–1 mg/L) [23,24], linezolid (0.5–1 mg/L) [25], and the first-line drug ethambutol (1–5 mg/L) [23]. However, FTSD2 MICs were lower than those of capreomycin (10 mg/L) or streptomycin (8 mg/L) [23], suggesting a promising antimicrobial activity profile.
The absence of cross-resistance with other drugs, such as rifampicin, isoniazid, and ethionamide, suggests that FTSD2 acts via a different mechanism of action. Additionally, the MBC was equivalent to the MIC for all tested strains, indicating that FTSD2 exhibits strong bactericidal activity. A previous in vitro study [24], reported that the MIC values of antitubercular drugs such as rifampicin, isoniazid, and levofloxacin were lower than those obtained for the MBC [24]. These results highlight FTSD2 potential to eliminate the bacillus in vitro, which could be of interest when considering its combination with other drugs. Additionally, FTSD2 showed no antibacterial activity against nontuberculous mycobacteria, such as M. avium, M. abscessus, M. fortuitum, M. chelonae, and M. smegmatis, confirming its in vitro antituberculosis specificity.
The in vitro time-kill evaluation showed that FTSD2 exerts a concentration-dependent bactericidal effect, achieving sterilizing activity at 192 h at a concentration of 8 mg/L (8×). This behavior is comparable to that of first-line antitubercular drugs, such as isoniazid, rifampicin, and ethambutol, which have shown concentration-dependent bactericidal effects in previous studies [26,27,28]. However, only rifampicin showed sterilizing activity at 144 h at a concentration of 2 mg/L (8× the drug’s MIC) [27], as observed in this study with FTSD2. Further studies are necessary to determine whether there is synergy between FTSD2 and other antitubercular drugs.
Cytotoxicity assays showed that FTSD2 did not induce toxicity in macrophages at concentrations below 64 mg/L. Higher concentrations (128 and 256 mg/L) were tested to assess the compound’s safety margin and determine the concentration threshold at which cytotoxic effects begin to appear. These assays demonstrated that the cell viability of hMDM and RAW 264.7 macrophages treated with FTSD2 remained above 94% after 48 h of exposure to a concentration of 256 mg/L. Previous studies, such as those reported in [29], revealed that the viability percentage of J774.A1 macrophages treated with rifampicin at a concentration of 10 mg/L was 88% [29]. This suggests that FTSD2 has a competitive advantage in terms of cell safety, especially considering its low toxicity in human and murine macrophages.
Since M. tuberculosis proliferates inside macrophages, developing drugs capable of eliminating intracellular mycobacteria is crucial for improving treatment outcomes [29]. FTSD2 was found to exhibit bactericidal activity in a macrophage model, showing prolonged inhibition of intracellular bacterial growth, comparable to that of rifampicin. Additionally, it was found that increasing the drug concentration resulted in a marked reduction in bacterial recovery, indicating greater penetration of the drug into macrophages [30]. The findings of this study suggest that FTSD2 could be integrated into future therapeutic regimens, particularly for multidrug-resistant tuberculosis.
While this study focuses on the in vitro evaluation of FTSD2, it lays the groundwork for future research, particularly in assessing its pharmacokinetics and pharmacodynamics in animal models to further evaluate its efficacy and safety against drug-susceptible and drug-resistant TB and its effectiveness in combination with other antitubercular drugs. Although no pharmacokinetic or animal toxicity studies have been conducted to date, future investigations are necessary to determine the compound’s pharmacokinetic profile, toxicity, and potential in vivo efficacy against drug-resistant tuberculosis. Additionally, it would be interesting to explore the exact mechanism of action of FTSD2 and its potential interactions with emerging resistant mutations, such as those associated with bedaquiline and delamanid. Ultimately, clinical trials are required to confirm its efficacy and safety in humans.
4. Materials and Methods
4.1. Strains
Seven drug-resistant clinical isolates of Mycobacterium tuberculosis were selected from the collection of the Reina Sofía University Hospital in Córdoba, Spain (three resistant to isoniazid, two resistant to isoniazid and rifampicin, and two resistant to isoniazid, rifampicin, and ethionamide), as well as the M. tuberculosis strains ATCC 35820, ATCC 35822, ATCC 35837, H37Rv, and H37Ra. Additionally, clinical isolates of M. avium, M. abscessus, M. chelonae, M. fortuitum, and the strain M. smegmatis mc^2^ 155 were included (Supplementary Material Table S4). Before starting the assays, all strains and clinical isolates were reactivated in Löwenstein−Jensen slant cultures for 3 to 4 weeks at 35 ± 1 °C for M. tuberculosis and M. avium, while the rapidly growing mycobacteria were incubated at 30 °C for 3 to 5 days.
4.2. Triaminopyrimidine-Diazepane Compounds
The synthesis of 4-(2,4-diamino-8-(p-tolyl)-8,9-dihydro-7H-pyrimido[4,5-b][1,4]diazepin-6-yl)-N-(2-((4-(dimethylamino)-6-((4-fluorophenyl)amino)-1,3,5-triazin-2-yl)amino)ethyl)benzenesulfonamide (Figure 4) was reported in a previous study [15]. The methodology employed is briefly described as follows: Initially, successive nucleophilic substitution reactions of 2,4,6-trichloro-1,3,5-triazine with 4-fluoroaniline, dimethylamine, and ethylenediamine were carried out, respectively, to obtain N2-(2-aminoethyl)-N4-(4-fluorophenyl)-N6,N6-dimethyl-1,3,5-triazine-2,4,6-triamine. This trisubstituted triazine was then reacted with 4-acetylbenzenesulfonyl chloride in the presence of triethylamine to obtain 4-acetyl-N-(2-((4-(dimethylamino)-6-((4-fluorophenyl)amino)-1,3,5-triazin-2-yl)amino)ethyl)benzenesulfonamide. Subsequently, using this carbonyl precursor as starting material, (E)-N-(2-((4-(dimethylamino)-6-((4-fluorophenyl)amino)-1,3,5-triazin-2-yl)amino)ethyl)-4-(3-(p-tolyl)acryloyl)benzenesulfonamide was obtained by Claisen–Schmidt condensation reactions with 4-methylbenzaldehyde in a basic medium. Finally, diazepine 1 was obtained by reacting (E)-N-(2-((4-(dimethylamino)-6-((4-fluorophenyl)amino)-1,3,5-triazin-2-yl)amino)ethyl)-4-(3-(p-tolyl)acryloyl)benzenesulfonamide with an excess of 2,4,5,6-tetraaminopyrimidine dihydrochloride and BF3·OEt2 in methanol under reflux.
4.3. Preparation of Stock Solutions
Due to the moderate aqueous solubility of FTSD2, DMSO was used as a solvent to prepare stock solutions. Stock solutions of FTSD2 and rifampicin were prepared in 100% DMSO at a concentration of 10 mg/mL. Before use in experimental assays, these solutions were diluted in the corresponding media to ensure that the final DMSO concentration in all tests did not exceed 0.1% (v/v). To eliminate any potential solvent-related interference, drug-free media were supplemented with 0.1% DMSO as a control.
4.4. Determination of Minimum Inhibitory Concentration (MIC) and Minimum Bactericidal Concentration (MBC)
The MIC and MBC were determined using the EUCAST broth microdilution method for the Mycobacterium tuberculosis complex [31]. Briefly, drug concentrations ranging from 8 mg/L to 0.015 mg/L were prepared in 96-well polystyrene U-bottom plates containing Middlebrook 7H9 medium supplemented with 10% Middlebrook OADC. As a growth control, 100 µL of drug-free medium containing 0.1% DMSO was added. Approximately 5–7 colonies from the Löwenstein−Jensen medium were vortexed in tubes with 3 mm glass beads (Merck, Darmstadt, Germany) and resuspended in 7 mL of distilled water. After 20 min of settling, the supernatant was adjusted to a McFarland standard of 0.5 (1.5 × 10^8^ CFU/mL) and diluted 1:50 to create an inoculum. A total of 100 µL of inoculum was added to each well. Plates were incubated at 35 ± 1 °C for 21 days for slow-growing mycobacteria and 5 days for rapidly growing species. The MIC was defined as the lowest concentration with no visible growth.
For MBC, 50 µL from wells without visible growth were plated on 7H10 medium supplemented with OADC. The plates were incubated at 35 ± 1 °C for 21 days. MBC was defined as the lowest drug concentration that reduced the initial inoculum by 99.9%. All experiments were performed in triplicate.
All experiments were performed in three independent biological replicates, each conducted in technical triplicates.
4.5. Kill Rate Determination
The killing rate of M. tuberculosis H37Rv exposed to FTSD2 was determined using a time-kill assay. A bacterial suspension adjusted to 0.5 McFarland units was prepared as described in the previous section. A total of 0.2 mL of this solution was added to flasks containing 9.8 mL of culture medium supplemented with the compound at concentrations of 1×, 2×, 4×, and 8× MIC, and a drug-free growth control. Serial dilutions (1:10) were performed for each condition up to a 10^−6^ dilution, and 10 µL aliquots were plated in triplicate on Petri dishes containing 7H10 agar medium supplemented with OADC at 0, 24, 48, 192, and 384 h. The plates were incubated at 36 ± 1 °C for 21 days, and colony counts were performed on dilutions where between 30 and 300 colonies were observed.
Time-kill assays were conducted in two independent experiments, each performed in triplicate.
4.6. Cytotoxicity Assay in Macrophages
The cytotoxicity of the FTSD2 compound was evaluated by its ability to eliminate murine RAW 264.7 macrophage cell lines and human monocyte-derived macrophages (hMDMs), using the macrophage-to-myofibroblast transition (MMT) method and the MTT colorimetric assay (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide), as described by [32].
4.6.1. MMT Assay
hMDMs were obtained from 50 mL of defibrinated blood from healthy donors, following a density gradient cell separation protocol. Blood was mixed at a 1:1 ratio with calcium- and magnesium-free DPBS (Dulbecco’s Phosphate Buffered Saline). The mixture was then centrifuged at 374× g for 20 min at 37 °C using Ficoll-Hypaque 1077 at a 1:3 ratio (blood) to separate the mononuclear cells. The obtained mononuclear cells were washed twice with DPBS and centrifuged again at 15.815× g for 10 min. Finally, the cells were resuspended in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% autologous serum, and the concentration was adjusted to 0.3 × 10^6^ cells/mL. The cells were incubated at 37 °C and 5% CO_2_ for 72 h in 24-well plates to promote differentiation into macrophages.
4.6.2. MTT Assay
The MTT assay was carried out in 96-well plates. Both hMDMs and murine RAW 264.7 macrophages were adjusted to a concentration of 2.5 × 10^4^ cells/mL in a supplemented DMEM medium. A total of 180 µL of the cell suspension was added to the wells, followed by 20 µL of FTSD2, to obtain concentrations ranging from 0.5 to 512 times the MIC (0.5 to 512 mg/L). Drug-free medium was used as a growth control. Doxorubicin was used as a positive cytotoxicity control because of its ability to eliminate both tumor and non-tumor cells. The plates were incubated for 48 h at 37 °C and 5% CO_2_. After incubation, the supernatant was removed from each well and the cells were washed with PBS. Fresh medium containing 0.5 mg/mL MTT was then added, and the cells were incubated for an additional 4 h at 37 °C. After incubation, the supernatant was removed, and 100 µL of pure DMSO was added to dissolve the formed crystals, and absorbance was measured at 540 nm using a spectrophotometer. The experiments were performed in triplicate, and statistical significance was determined using one-way ANOVA followed by Tukey’s post hoc test.
Cell viability was calculated using MTT reduction, and cytotoxicity was defined as a decrease in cell viability compared to that of untreated cells. The formula used was: (%) viability inhibition = 1 − [(OD_540nm_ treated cells/OD_540nm_ growth control) × 100], where the absorbance of the growth control represents 100% viability.
Cytotoxicity assays were performed in two independent experiments, each conducted in technical triplicates.
4.7. Macrophage Assay
The intracellular activity of FTSD2 was assessed using a macrophage infection model as previously described by Khara et al. (2016) [33] with slight modifications. Briefly, RAW 264.7 cells were seeded at 2.5 × 10^4^ cells per well in 96-well plates and incubated for 24 h to allow adherence. Prior to infection, M. tuberculosis H37Rv cultures were washed twice with PBS, resuspended in DMEM, and added at a final concentration of 3 × 10^5^ cfu per well to achieve an moi of 10:1. After a 4-h incubation at 37 °C and 5% CO_2_, extracellular bacteria were removed by washing with DMEM. Cells were treated with FTSD2 or rifampicin at 1× and 4× MIC. Macrophages were lysed using sterile water for 30 min at different time points (0, 24, 48, 72, 96, 120, and 148 h), and bacterial viability was assessed by plating on 7H10 agar plates. The experiments were performed in triplicate, and statistical significance was determined using one-way ANOVA followed by Tukey’s post hoc test. The results are expressed as log CFU/mL.
Intracellular bacterial growth inhibition assays were performed in two independent experiments, each conducted in technical triplicates.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1World Health Organization Global Tuberculosis Report 2024 WHO Geneva, Switzerland 2024
- 2Cohen J. Approval of Novel TB Drug Celebrated—With Restraint Science (1979)201333913010.1126/science.339.6116.13023307714 · doi ↗ · pubmed ↗
- 3Ryan N.J. Lo J.H. Delamanid: First Global Approval Drugs 2014741041104510.1007/s 40265-014-0241-524923253 · doi ↗ · pubmed ↗
- 4Thakare R. Dasgupta A. Chopra S. Pretomanid for the Treatment of Pulmonary Tuberculosis Drugs Today 20205665510.1358/dot.2020.56.10.316123733185630 · doi ↗ · pubmed ↗
- 5World Health Organization WHO Consolidated Guidelines on Tuberculosis. Module 4: Treatment—Drug-Susceptible Tuberculosis Treatment World Health Organization Geneva, Switzerland 202235727905 · pubmed ↗
- 6World Health Organization Global Tuberculosis Report 2023 World Health Organization Geneva, Switzerland 20239789240083851
- 7Moe S. Rekart M.L. Hernandez D. Sholpan A. Ismailov A. Oluya M. Bayniyazova A. Zinaida T. Nargiza P. Gomez-Restrepo C. Primary Bedaquiline Resistance in Karakalpakstan, Uzbekistan Int. J. Tuberc. Lung Dis.20232738138610.5588/ijtld.22.053637143220 PMC 10171487 · doi ↗ · pubmed ↗
- 8Huitric E. Verhasselt P. Koul A. Andries K. Hoffner S. Andersson D.I. Rates and Mechanisms of Resistance Development in Mycobacterium Tuberculosis to a Novel Diarylquinoline ATP Synthase Inhibitor Antimicrob. Agents. Chemother.2010541022102810.1128/AAC.01611-0920038615 PMC 2825986 · doi ↗ · pubmed ↗