Genetic Variations of Three Kazakhstan Strains of the SARS-CoV-2 Virus
Bekbolat Usserbayev, Kulyaisan T. Sultankulova, Yerbol Burashev, Aibarys Melisbek, Meirzhan Shirinbekov, Balzhan S. Myrzakhmetova, Asankadir Zhunushov, Izat Smekenov, Aslan Kerimbaev, Sergazy Nurabaev, Olga Chervyakova, Nurlan Kozhabergenov, Lesbek B. Kutumbetov

TL;DR
This study sequenced three SARS-CoV-2 strains from Kazakhstan and identified their genetic variations and mutations.
Contribution
The paper provides new insights into the genetic diversity of SARS-CoV-2 strains circulating in Kazakhstan.
Findings
Three SARS-CoV-2 strains were sequenced, revealing 127 mutations compared to the reference strain.
Common mutations like D614G were found in all three strains, suggesting potential functional significance.
Phylogenetic analysis showed the strains are related to samples from Germany and Mexico.
Abstract
Prompt determination of the etiological agent is important in an outbreak of pathogens with pandemic potential, particularly for dangerous infectious diseases. Molecular genetic methods allow for arriving at an accurate diagnosis, employing timely preventive measures, and controlling the spread of the disease-causing agent. In this study, whole-genome sequencing of three SARS-CoV-2 strains was performed using the Sanger method, which provides high accuracy in determining nucleotide sequences and avoids errors associated with multiple DNA amplification. Complete nucleotide sequences of samples, KAZ/Britain/2021, KAZ/B1.1/2021, and KAZ/Delta020/2021 were obtained, with sizes of 29.751 bp, 29.815 bp, and 29.840 bp, respectively. According to the COVID-19 Genome Annotator, 127 mutations were detected in the studied samples compared to the reference strain. The strain KAZ/Britain/2021…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1 Figure 2
Figure 2 Figure 3
Figure 3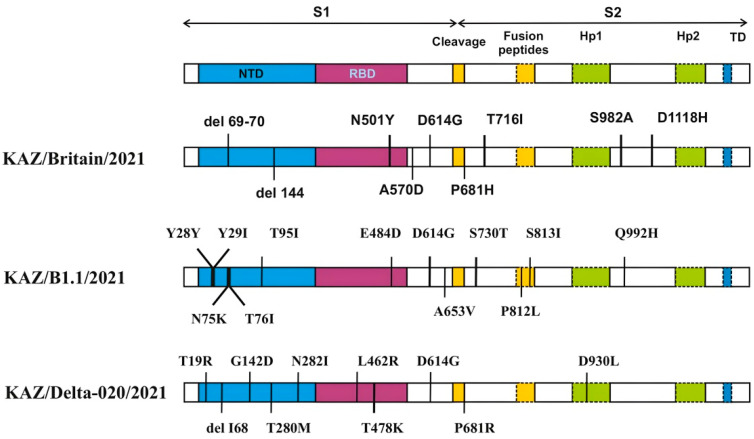 Figure 4
Figure 4 Figure 5
Figure 5- —Development of a vaccine against coronavirus infection COVID-19
- —Science Committee of the Ministry of Education and Science of the Republic of Kazakhstan
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSARS-CoV-2 and COVID-19 Research · Viral gastroenteritis research and epidemiology · Bacillus and Francisella bacterial research
1. Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent of coronavirus disease 2019 (COVID-19), was first reported in December 2019 in Wuhan, Hubei Province, China [1]. According to WHO COVID-19 data, as of 23 February 2025, a total of 777,519,152 confirmed cases have been reported, of which 7,090,776 have resulted in death [2]. The first cases of COVID-19 in the Republic of Kazakhstan were reported on 13 March 2020 [3,4].
The SARS-CoV-2 virion is spherical or ellipsoidal, with an average diameter ranging from 60 to 140 nanometers [5]. The SARS-CoV-2 virus genome consists of ~29.9 kb and is organized in the following order from 5′ to 3′: open reading frame (ORF) 1ab (replicase), structural spike glycoprotein (S), ORF3a protein, structural envelope protein (E), structural membrane glycoprotein (M), ORF6 protein, ORF7a protein, ORF7b protein, ORF8 protein, structural nucleocapsid phosphoprotein (N), and ORF10 protein [6]
Over time, all viruses, including SARS-CoV-2, undergo molecular genetic changes. Most of these changes have little effect on the properties of the virus. However, some mutations can affect various aspects, such as its infectivity, transmissibility, the effectiveness of treatment and vaccines, as well as virulence [7]. In addition, since its emergence in 2019, the SARS-CoV-2 virus has undergone continuous changes, which contributed to the emergence of multiple lineages and variants (Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1), Delta (B.1.617.2), and Omicron (B.1.1.529)) [8,9], having differences in transmission characteristics, ability to cause severe disease, and ability to evade immune response [10].
The expansion of the complete genomic sequences of the SARS-CoV-2 virus in the information databases (GISAID and GenBank NCBI) was made possible by rapid genome sequencing using Sanger or NGS methods [11]. These SARS-CoV-2 genomes in the context of the COVID-19 pandemic can provide invaluable information on the evolution of the virus and allow tracking of the geographic distribution of individual mutations, as well as monitoring of the spread of the virus in the human population [12]. In addition, the evolution of the virus is facilitated by the adaptation of the virus in different conditions and results from a balance between its genetic information and genome variability [13]. Studying the evolution and genetic changes in the genomes of various variants of SARS-CoV-2 is extremely important in developing clinical and political strategies within geographical regions [14] as well as for the creation of diagnostic tests and vaccines against this virus [15].
The aim of this work is to sequence the complete genome of three isolates of the SARS-CoV-2 virus, determine their genetic variations, and identify various types of mutations present in the different strains.
2. Materials and Methods
2.1. Sample Collection
Three strains of the SARS-CoV-2 virus were received for molecular genetic studies at the Research Institute for Biological Safety Problems in 2022 from the Scientific and Practical Center for Sanitary and Epidemiological Expertise and Monitoring branch of the National Center for Public Health, a republican state enterprise on the right of economic use of the Ministry of Health of the Republic of Kazakhstan.
2.2. RNA Extraction
Total RNA was extracted from virus-containing fluid using the QIAamp Viral RNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. RNA concentrations were estimated using Qubit RNA HS assay kits (Life Technologies, Carlsbad, CA, USA) on a Qubit 2.0 fluorometer (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s protocol.
2.3. cDNA Synthesis
Reverse transcription (RT) was performed using the SuperScript VILO cDNA Synthesis Kit (Invitrogen, Thermo Fisher Scientific, Carlsbad, CA, USA) in a Mastercycler X50 s thermal cycler at the following conditions: 25 °C for 10 min; 42 °C for 60 min; 85 °C for 5 min. The reaction composition and temperature–time conditions were followed according to the manufacturer’s instructions.
2.4. Primer Design and Synthesis
Specific overlapping primers for amplification and sequencing of all SARS-CoV-2 virus genes were manually searched and designed on the NCBI website using the GenBank database. The nucleotide sequence of the sequencing primers was designed based on the SARS-CoV-2 isolate Wuhan-Hu-1 reference strain (NC_045512.2) [16]. The specificity of the primers was checked using NCBI Primer-BLAST [17]. The primers were designed so that each pair overlapped each other, and their sequences were conserved in all SARS-CoV-2 virus variants. As a result, 65 pairs of sequencing primers were selected to amplify the complete genome of SARS-CoV-2 virus variants with an overlap of about 100 nucleotide base pairs (bp). The estimated length of the amplicons ranged from 600 to 772 bp [18]. Oligonucleotides were synthesized on an automatic DNA/RNA Synthesizer H-16 oligonucleotide synthesizer (K&A Labs GmbH, Schaafheim, Germany) using the phosphoramidite method performed according to the manufacturer’s protocol. The synthesized primers were eluted from the columns with a concentrated ammonia solution. The primers were then dried on a rotary evaporator and purified by alcohol precipitate.
2.5. Polymerase Chain Reaction (PCR) Setup
Amplification was performed on a Mastercycler X50 s thermal cycler using the Platinum SuperFi PCR Master Mix kit (Invitrogen, Thermo Fisher Scientific, Vilnius, Lithuania) according to the manufacturer’s instructions. PCR was performed in a total volume of 25 µL, composed of: 12.5 µL of 2X Platinum SuperFi PCR Master Mix, 1.25 µL of each of 10 µM forward and reverse primers, 3 µL of cDNA template, 5 µL of 5X SuperFi GC Enhancer, and PCR-grade water to bring the volume to 25 µL. PCR products were amplified using the following conditions: initial denaturation 95 °C—0.5 min; with subsequent 35 amplification cycles with denaturation at 95 °C for 0.1 min, annealing at 57 °C for 0.5 min, elongation at 72 °C for 0.5 min; final elongation at 72 °C for 5 min.
Horizontal gel electrophoresis was performed in 1.5% agarose gel (TopVision Agarose, Thermo Fisher Scientific Baltics, UAB, Vilnius, Lithuania) stained with ethidium bromide in Tris-acetate buffer at a voltage of 100 volts/cm of gel length for 30 min. The gel was subsequently viewed using a MiniBIS Pro transilluminator (DNR Bio Imaging Systems, Ltd., Jerusalem, Israel). Visualization and documentation of gel electrophoresis results were performed using the GelCapture program (DNR Bio-Imaging Systems Ltd., Ha-Satat St, Modi’in-Maccabim-Re’ut, Israel). A 100 bp DNA Ladder (New England Biolabs, Ipswich, MA, USA) was used as a molecular mass marker. The PCR product was purified using the GeneJET PCR Purification Kit (Thermo Fisher Scientific, Carlsbad, CA, USA) according to the manufacturer’s instructions.
2.6. Determination of Nucleotide Sequences
Sequencing of the whole SARS-CoV-2 virus genome after purification of the PCR product was carried out using termination dideoxynucleotides (Sanger method) with the AB BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Inc., Austin, TX, USA) and specific overlapping primers designed from different viral genes used in the amplification step. The products were purified using the BigDye Xterminator kit (Applied Biosystems, Foster City, CA, USA) and sequenced using a 3130 XL Genetic Analyzer (HITACHI, Tokyo, Japan). After sequencing, the obtained nucleotide sequence data were processed using the Sequencher v.5.4 program (Gene Codes Corporation, Ann Arbor, MI, USA).
2.7. Lineage Determination and Mutation Identification of the Studied Isolates
The SARS-CoV-2 virus strain lineage determination was performed using the Pangolin COVID-19 database [19]. The alignment of the SARS-CoV-2 virus nucleotide sequences with the reference strain and the identification of mutations were performed using the COVID-19 Genome Annotator Tool and Annotator [20].
2.8. Analysis of Non-Synonymous Mutation Function
PROVEAN v1.1 software was used to determine whether the selected mutations independently resulted in potential loss of function or a neutral effect. Mutation scores above the default threshold of −2.5 imply a neutral effect, while scores below this threshold indicate a deleterious effect [21].
2.9. Phylogenetic Analysis of Nucleotide Sequences
Evolutionary analysis was performed in MEGA 11 [22]. A phylogenetic tree including three samples, a reference genome, and genomes of different SARS-CoV-2 lineages was constructed using the Neighbor-Joining method [23]. The percentage of replicates in which related taxa were grouped together in a bootstrap test (1000 replicates) was shown next to the branches [24]. The tree was drawn to scale, with branch lengths (next to the branches) in the same units as the evolutionary distances used to construct the phylogenetic tree. Evolutionary distance was calculated using the maximum composite likelihood method [25] and expressed in units of base substitutions per site. To construct the phylogenetic tree, sequences were first aligned in the GenBank database and viral nucleotide sequences that were similar to the sequence of the strain under study were selected. The most suitable substitution model was selected for tree construction. A preliminary tree was constructed using the appropriate model. Then, the tree was pruned, typical strains were selected by year and territory, and the lineage of the strain under study was determined. When constructing a new phylogenetic tree, strains of another lineage were selected.
3. Results
3.1. PCR Amplification of SARS-CoV-2 Virus Strains
After RNA extraction and cDNA synthesis, amplification was performed using specific sequencing primers [18] for the complete SARS-CoV-2 virus genome by PCR according to the manufacturer’s protocol described above. Figure 1, Figure 2 and Figure 3 show the results of the electropherogram using the developed 65 pairs of sequencing primers.
As can be seen in Figure 1, Figure 2 and Figure 3, fragments of the complete genome of the SARS-CoV-2 virus samples were obtained using the developed sequencing primers [18]. Electrophoretic analysis yielded products with a molecular weight ranging between 612–732 bp. The length of the obtained amplicons corresponds to the length of the synthesized sequencing primers.
3.2. Characteristics of the Genomes of the Studied SARS-CoV-2 Virus Strains
The size of the genomes of the studied samples SARS-CoV-2/human/KAZ/Britain/2021 (KAZ/Britain/2021), SARS-CoV-2/human/KAZ/B1.1/2021 (KAZ/B1.1/2021), and SARS-CoV-2/human/KAZ/Delta-020/2021 (KAZ/Delta020/2021) were 29.751 bp, 29.815 bp, and 29.840 bp, respectively, and the GC contents were 38%, 37.95%, and 38%, respectively [4,26,27].
The nucleotide sequences of Kazakhstan SARS-CoV-2 virus strains were analyzed using the Pangolin COVID-19 database [19]. According to Pangolin COVID-19 data, the studied strains KAZ/Britain/2021, KAZ/B1.1/2021, and KAZ/Delta020/2021 belong to the B.1.1.7, B.1.1, and AY.122 lineages of the SARS-CoV-2 virus, respectively.
The COVID-19 Genome Annotator tool [20] was used to detect mutations in the obtained nucleotide sequences. According to the COVID-19 Genome Annotator data, a total of 127 mutations were detected in the studied strains compared to the reference strain. The most variable regions in the analysis of the genomic distribution of SNP (Single Nucleotide Polymorphism) and amino acid substitutions were the ORF1ab protein, which makes up 2/3 of the SARS-CoV-2 virus (Table 1), and the S protein (Table 2 and Figure 4).
The data presented in Table 1 show that the analysis of mutations in the 5′UTR (untranslated region), ORF1ab and 3′UTR regions of the studied isolates revealed a total of 61 variations at the nucleotide level. Among them, 18 nucleotide substitutions and 1 deletion were found in the strain KAZ/Britain/2021, 21 nucleotide substitutions and 1 deletion in the strains KAZ/B1.1/2021, and 20 mutations in the strain KAZ/Delta020/2021. A mutation at position 241 in the 5′UTR region of the virus was detected in all three strains studied and resulted in a C to T nucleotide substitution. A C to T nucleotide substitution at positions 3037 and 14,408 in the ORF1ab region was detected in all strains studied and resulted in one silent substitution (SNP silent) at position 106 (F106F) and one missense mutation at position 314 (P314L), respectively. A deletion of one amino acid residue S106 (serine → deletion) was observed in two samples studied (KAZ/Britain/2021 and KAZ/B1.1/2021) in the NSP6 region.
Table 2 and Figure 4 show the distribution of SNP and amino acid substitutions identified in the S protein of the studied strains. A total of 31 mutations were found in the S protein of the studied strains. Among them, 7 amino acid substitutions and 2 deletions were detected in the KAZ/Britain/2021 strain, 12 amino acid substitutions in the KAZ/B1.1/2021 strain, and 9 amino acid substitutions and 1 deletion in the KAZ/Delta020/2021 strain.
Mutational changes in the virus occurred more often in the S1 region than in the S2 region. In the S1 region of the KAZ/Britain/2021 strain, 2 deletions in the NTD region and 4 amino acid changes were detected, of which 1 mutation belongs to the RBD region. In the S2 region, 3 amino acid changes were detected compared to the reference strain. In the strain KAZ/B1.1/2021, 12 mutations were detected compared to the original strain, such as Y28Y, T29I, N74K, T76I, T95I, E484D, D614G, A653V, S730T, P812L, S813I, and Q992H. In the S protein of the strain KAZ/Delta020/2021, sets of mutations (L452R, T478K, and P681R) were found, which are unique only to the Delta variant.
The distribution of SNPs and amino acid substitutions, in addition to the ORF1ab and S proteins, was observed in the ORF8, N, ORF7a, ORF3a, M, ORF6, and ORF7b proteins of the studied strains and amounted to 9, 9, 6, 4, 3, 2, and 1 amino acid substitution, respectively (Table 3).
As shown in Table 3, a total of 34 variations were detected in the three isolates compared to the original strain. In the current study, four mutations were detected in the ORF3a protein across different samples: one mutation (W149L) in the KAZ/Britain/2021 sample, two mutations (A99V, P240S) in the KAZ/B1.1/2021 strain, and one mutation (S26L) in the KAZ/Delta020/2021 strain. Three mutations were detected in the M protein across the strains: two mutations (H125Y and K162N) in the KAZ/B1.1/2021 strain and one mutation (I82T) in the KAZ/Delta020/2021 strain. Two mutations (W27 and NL28KF) were detected in the ORF6 protein, which were found only in the KAZ/Britain/2021 strain. Six mutations were detected in the ORF7a protein: three mutations (A79A, E92K and L116F) were detected in the KAZ/B1.1/2021 strain, and three mutations (P45L, V82A and T120I) were detected in the KAZ/Delta020/2021 strain. Only one mutation (T40I) was detected in the ORF7b protein in the KAZ/Delta020/2021 strains. Eight mutations were detected in the ORF8 protein: four mutations (Q27, R52I, K68, and Y73C) were found in the KAZ/B1.1/2021 strain, and four mutations (F120L, F120L, I212N, and 122) were found in the KAZ/Delta020/2021 strain. Nine mutations were detected in the N protein compared to the original virus strain: three mutations in the KAZ/Britain/2021 strain (D3L, RG203KR and S235F), two mutations in the KAZ/B.1.1/2021 strain (RG203K and K388I), and four mutations in the KAZ/Delta020/2021 strain (D63G, R203M, G215C, G312G, and D377Y).
3.3. Impact of Mutations on Biological Function of Proteins in the Studied SARS-CoV-2 Samples
The PROVEAN web server was used to assess whether the selected mutations could lead to a potential loss of function or remain neutral. Loss of function occurs when a mutation leads to the formation of a non-functional protein. At the same time, a neutral result means that the protein function is preserved despite the presence of a mutation. The PROVEAN platform is focused only on the analysis of the individual effects of each of the mutations identified in the studied virus isolates (Table 4) [28].
Table 4 shows that the proportion of loss-of-function mutations detected in the three studied genomes (KAZ/Britain/2021, KAZ/B1.1/2021, and KAZ/Delta020/2021) of SARS-CoV-2 was studied, and 5 (P218L, T716I, W149L, R52I, and Y73C), 2 (S813I, and Q992H), and 8 (P77L, L452R, I82T, P45L, V82A, F120L, F120L, and R203M) loss-of-function mutations were identified, respectively. Among the genes in the studied samples, the proportion of loss-of-function mutations was higher in the S and ORF8 genes than in other genes.
3.4. Phylogenetic Analysis
Phylogenetic analysis between the studied isolates and other isolates belonging to different lineages of the SARS-CoV-2 virus from the international GenBank NCBI database are presented in Figure 5.
Based on the phylogenetic analysis, the studied strains KAZ/Britain/2021, KAZ/B1.1/2021 and KAZ/Delta020/2021 belong to different SARS-CoV-2 lineages. KAZ/Britain/2021 formed a group (bootstrap (BS) = 100%) with isolates belonging to the B.1.1.7 SARS-CoV-2 lineage. The nucleotide identity between them ranged from 99.96 to 99.97 percent. Within the monophylogenetic group, OU141323.1 SARS-CoV-2/Germany/2021 was the most similar to KAZ/Britain/2021, with a nucleotide similarity of 99.97%. KAZ/B1.1/2021 groups (bootstrap (BS) = 100%) with various samples belonging to the B.1.1 SARS-CoV-2 lineage. KAZ/B1.1/2021 closely matched the samples from Mexico (OK435605.1), showing a nucleotide identity of 99.84%. KAZ/Delta020/2021 formed a monophyletic group (bootstrap (BS) = 61% and (BS) = 100%) with samples that belong to the AY.122 and B.1.617.2 lineages, respectively. However, our KAZ/Delta020/2021 showed high similarity to isolates from Germany (OV375251.1 and OU975174.1), which had a nucleotide identity of 99.94%.
4. Discussion
Cleaveland S. et al. reported that most viruses infecting humans are zoonotic. Zoonotic viruses, after entering a cell, adapt inefficiently to a new host and replicate and transmit slowly [29]. Their transmission from animal to human and from human to human depends on many factors, including potential adaptive evolution to virulent strains [30].
RNA viruses are characterized by higher replication fidelity (∼10^−4^ error/site/cycle) and genetically diverse RNA polymerases [31]. However, when RNA viruses circulate in the community, genetic changes continuously occur due to copying errors of RNA polymerase. This, in turn, leads to mutations in the genome [32]. Lee et al. analyzed the rate of genome evolution of several SARS-CoV-2 virus strains over one month and found that the average evolution rate ranged from 1.7926 × 10^−3^ to 1.8266 × 10^−3^ substitutions per site per year [33,34], but four months after the pandemic, the mutation rate of the whole SARS-CoV-2 virus genome was 3.95 × 10^−4^ per nucleotide per year [35].
The rapid evolution of the SARS-CoV-2 genome highlights the need to develop antiviral drugs against the virus [36]. To develop effective antiviral drugs, it is necessary to determine which variant is most actively circulating in society during the pandemic. This depends on the data collected on COVID-19 infection, the epidemiological features among different population groups, as well as the patterns of viral spread in different areas. The modern approach to the use of genomic and information technologies in epidemiological surveillance of SARS-CoV-2 pathogens occupies an important place in measures to prevent and control the virus [37].
Sanger sequencing is considered the most optimal method for sequencing short fragments (<1000 bp) and is useful for filling gaps in partial whole genomes [38,39]. An important step for the successful implementation of the Sanger method is the production of a PCR amplicon from the samples under study and the development of sequencing oligonucleotide primers for the amplification of this PCR amplicon [32].
It is impossible to obtain the complete genomic nucleotide sequence of SARS-CoV-2 virus in a single reaction using the Sanger method. Therefore, in our current study, we designed a set of sequencing primers targeting SARS-CoV-2 virus to obtain the complete genomic nucleotide sequence [18]. The specific designed sequencing primers were selected based on the Wuhan-Hu-1 reference strain, and each designed primer pair overlapped with each other and their sequence was conserved among all SARS-CoV-2 virus variants. The length of the sequencing primers ranged from 600 bp to 772 bp with a GC content of 38% to 50%, and the melting temperature was in the range of 55–57 °C. The PCR products of the studied samples were obtained using the designed sequencing primers.
The length of the amplicons obtained (Figure 1, Figure 2 and Figure 3) corresponds to the length of the synthesized sequencing primers. The developed specific primers covered 100% and amplified the entire genome of the studied samples. After sequencing, the nucleotide sequences of the studied samples were obtained and analyzed in the Pangolin COVID-19 database [19]. According to the Pangolin COVID-19 data, the KAZ/Britain/2021 strain belongs to the B.1.1.7 lineage (Alpha variant). Alpha differs from other variants of the virus by the presence of mutations in the S protein, such as deletion 69–70, deletion 144, N501Y, A570D, D614G, P681H, T716I, S982A, and D1118H [40,41]. The mutations identified in the S gene of the studied isolate KAZ/Britain/2021 are 100% consistent with the mutations found in the S gene of the alpha variant (Table 2 and Figure 4).
Bo Meng et al. suggest that the H69-V70 deletion in the NTD region of the S1 subunit of the spike protein, found in the studied SARS-CoV-2 virus sample, is associated with increased infectivity and evasion of the host immune response [42,43,44]. Weng S. et al. confirmed that the Δ144/145 deletion blocks the binding sites of neutralizing antibodies, which is important in preventing the virus from entering the cell and possibly interfering with its replication [44,45]. Some studies describe H69-V70, N501Y, and P681H, which may affect viral infectivity [42,46]. N501Y increases viral infectivity by 70–80% and enhances the binding affinity of the viral S protein to human ACE2 [42,46,47,48]. According to some studies, mutations A570D, T716I, S982A, and D1118H are the result of accumulated mutations of the virus in the community environment, which together increase the lethality and transmissibility of the SARS-CoV-2 virus [48,49]. D614G was found in all three isolates and has become the most common mutation among SARS-CoV-2 variants during the global pandemic [50]. Lubinski B. et al. showed that P681H can increase its cleavage by furin-like proteases, although this process does not lead to viral entry [51]. According to Pangolin COVID-19 data, strain KAZ/B1.1/2021 belongs to the B.1.1 lineage. Currently, the SARS-CoV-2 virus is divided into two lineages: A and B. Lineage B includes 47 different lineages, and Lineage B.1.1 is part of this lineage [52,53,54].
The strain KAZ/Delta020/2021, according to Pangolin COVID-19, belongs to the AY.122 lineage (Delta variant, B.1.617). As indicated by the source SARS-CoV-2 Lineage Tree, B.1.617 is divided into three sublineages: B.1.617.1, B.1.617.2, and B.1.617.3. Dhawan M. et al. note that the B.1.617.2 lineage emerged during the second wave of coronavirus infection in India. The B.1.617.2 lineage includes 134 different lineages, one of which is AY.122. Some literature sources emphasize that B.1.617.2 is characterized by differences from other viral variants due to a unique set of mutations, such as L452R, T478K, and P681R. These mutations make it particularly infectious and resistant to neutralizing antibodies in previously infected or vaccinated individuals [55,56,57,58]. Other studies have also shown that the T19R, T478K, P681R, and D950N mutations found in the S gene enhance viral replication and help it evade the host’s immune response [59,60].
The resulting nucleotide sequences were tested using the COVID-19 Genome Annotator [20] to detect mutations. According to the COVID-19 Genome Annotator, a total of 127 mutations were detected in the isolates tested compared to the reference strain. In these studied isolates, the following types of mutations were encountered: SNP, silent mutations, stop codon, deletion and mutations occurring in the 5′ and 3′ untranslated region in the genome compared to the original strain, and their quantities in the studied genomes were 92, 17, 3, 5, 5, and 5, respectively (Table 1, Table 2 and Table 3 and Figure 4). Analysis of the distribution of SNP in the studied genomes showed that the most common in the ORF1ab gene (n = 36) and S (n = 27). The main silent mutations were found in the ORF1ab gene (n = 13), in the remaining genes S, ORF7b, ORF8 and N only one was detected, respectively. Deletions were found only in the S gene (n = 3) and ORF1ab (n = 2).
The study identified mutations in the 5′UTR (C241T), ORF1ab (F106F and P314L) and S (D614G) regions that were common to all three isolates studied. The study by Periwal N et al. showed that the C nucleotide at position 241 in the 5′UTR region was replaced by a T nucleotide as early as the summer of 2020 [61]. Kim et al. reported that this mutation in the 5′UTR region may affect the rate of transcription and replication of the SARS-CoV-2 virus [12,62,63]. Some studies predicted that the synonymous F106F mutation identified in the NSP3 region of the ORF1ab gene may play a role in mRNA processing, altering the properties of the viral protein [12,63]. The missense mutation P314L, found in the NSP12 region of the ORF1b gene, is considered to be part of the core replication/transcription complex and is a conserved protein in coronaviruses [6,64]. Thus, the P314L mutation affects SARS-CoV-2 RNA replication by participating in the activity of RdRp (RNA-dependent RNA polymerase) [65,66]. In addition, RdRp plays an important role in the process of SARS-CoV-2 viral replication and transcription [67]. D614G was detected in all three isolates and has become the most common mutation among SARS-CoV-2 variants during the global pandemic [50].
It is important to evaluate the change in function of the mutations identified in the study, which may have effects on viral circulation. Aside from this, further studies on these mutations can contribute to the development of various antiviral drugs against SARS-CoV-2. This study revealed significant changes in amino acids in structural and accessory proteins (P218L and P77L in ORF1ab; T716I, S813I, Q992H, and N282I in S; W149L in ORF3a; I82T in M; P45L and V82A in ORF7a; R52I, Y73C, and F120L in ORF8; R203M in N), which may cause functional alterations and affect functional characteristics of the virus.
Phylogenetic analysis of SARS-CoV-2 virus isolates showed that the studied samples belong to different virus lineages. In the study, the KAZ/Britain/2021 strain showed significant similarity to the OU141323.1 SARS-CoV-2/Germany/2021 isolate and formed a group with strains that belong to the B.1.1.7 lineage. The nucleotide similarity between these isolates was 99.97%, indicating their very close genetic relationship. KAZ/B1.1/2021 grouped with various isolates that belong to the B.1.1 lineage. In addition, it showed close similarity to samples obtained from Mexico (OK435605.1) SARS-CoV-2/human/MEX/CMX-51/2020), demonstrating a nucleotide identity of 99.84%. KAZ/Delta020/2021 clustered with isolates belonging to the AY.122 and B.1.617.2 lineages. However, KAZ/Delta020/2021 showed high similarity to isolates from Germany—OV375251.1 and OU975174.1—with 99.94% nucleotide identity. According to Pangolin COVID-19 data, the AY.122 lineage is one of the sublineages of B.1.617. B.1.617 emerged in late 2020 in Maharashtra, India [68]. In mid-June 2021, a mutated Delta variant (B.1.617.2), known as the Delta plus, was identified in India [69].
Therefore, the SARS-CoV-2 samples were fully amplified and sequenced using the developed primers, which allowed us to identify mutations compared to the reference strain Wuhan-Hu-1 (NC_045512.2). Sanger-based whole-genome sequencing of the studied SARS-CoV-2 isolates was successfully demonstrated. The data obtained using molecular genetic methods during the pandemic are of great importance for understanding the biology of the virus, developing new diagnostic and therapeutic methods, and making informed public health decisions. Continued research in this area will allow us to be better prepared for future pandemics.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Shereen M.A. Khan S. Kazmi A. Bashir N. Siddique R. COVID-19 infection: Origin, transmission, and characteristics of human coronaviruses J. Adv. Res.202024919810.1016/j.jare.2020.03.00532257431 PMC 7113610 · doi ↗ · pubmed ↗
- 2World Health Organization COVID-19 Dashboard Available online: https://covid 19.who.int/(accessed on 6 March 2025)
- 3Zhugunissov K. Zakarya K. Khairullin B. Orynbayev M. Abduraimov Y. Kassenov M. Sultankulova K. Kerimbayev A. Nurabayev S. Myrzakhmetova B. Development of the Inactivated Qaz Covid-in Vaccine: Protective Efficacy of the Vaccine in Syrian Hamsters Front. Microbiol.20211272043710.3389/fmicb.2021.72043734646246 PMC 8503606 · doi ↗ · pubmed ↗
- 4Usserbayev B. Zakarya K. Kutumbetov L. Orynbayev M. Sultankulova K. Abduraimov Y. Myrzakhmetova B. Zhugunissov K. Kerimbayev A. Melisbek A. Near-complete genome sequence of a SARS-Co V-2 variant B. 1.1. 7 virus strain isolated in Kazakhstan Microbiol. Resour. Announc.202211 e 006192210.1128/mra.00619-2235997492 PMC 9476996 · doi ↗ · pubmed ↗
- 5Zhu N. Zhang D. Wang W. Li X. Yang B. Song J. Zhao X. Huang B. Shi W. Lu R. A novel coronavirus from patients with pneumonia in China, 2019 N. Engl. J. Med.202038272773310.1056/NEJ Moa 200101731978945 PMC 7092803 · doi ↗ · pubmed ↗
- 6Wu F. Zhao S. Yu B. Chen Y.M. Wang W. Song Z.G. Hu Y. Tao Z.W. Tian J.H. Pei Y.Y. A new coronavirus associated with human respiratory disease in China Nature 202057926526910.1038/s 41586-020-2008-332015508 PMC 7094943 · doi ↗ · pubmed ↗
- 7Cosar B. Karagulleoglu Z.Y. Unal S. Ince A.T. Uncuoglu D.B. Tuncer G. Kilinc B.R. Ozkan Y.E. Ozkoc H.C. Demir I.N. SARS-Co V-2 Mutations and their Viral Variants Cytokine Growth Factor. Rev.202263102210.1016/j.cytogfr.2021.06.00134580015 PMC 8252702 · doi ↗ · pubmed ↗
- 8Safari I. Elahi E. Evolution of the SARS-Co V-2 genome and emergence of variants of concern Arch. Virol.202216729330510.1007/s 00705-021-05295-534846601 PMC 8629736 · doi ↗ · pubmed ↗