Primary Pyrrolimines and Pyridinimines
Amavi Kpoezoun, Gnon Baba, Jean-Claude Guillemin

TL;DR
This paper explores the synthesis and properties of new imine compounds linked to pyrrole or pyridine rings.
Contribution
The study introduces and isolates new pyrrolimines and pyridinimines with N-unsubstituted imine groups.
Findings
Pyrrole and pyridine derivatives with N-unsubstituted imine groups were successfully synthesized.
These compounds are less volatile and more challenging to isolate compared to similar aromatic derivatives.
The work opens new avenues for understanding the physicochemical properties of these understudied compounds.
Abstract
The association of an aromatic ring with an N-H-unsubstituted imine generates families of compounds that have been little studied until now except when the ring is a phenyl group. Recently, such imines substituted by a furan or thiophene group have been synthesized. This work reports a similar study where a pyrrole or pyridine ring is directly linked to an N-unsubstituted aldimine or ketimine group in order to isolate such compounds and to open the way to the knowledge of their physicochemical properties. The lower volatility of pyrrole and pyridine derivatives compared to aryl, furan, or thiophene derivatives greatly increases the difficulty of the synthesis and isolation of these kinetically unstable compounds.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
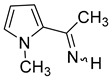 Figure 1
Figure 1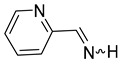 Figure 2
Figure 2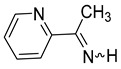 Figure 3
Figure 3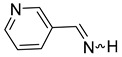 Figure 4
Figure 4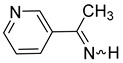 Figure 5
Figure 5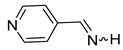 Figure 6
Figure 6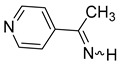 Figure 7
Figure 7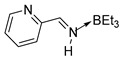 Figure 8
Figure 8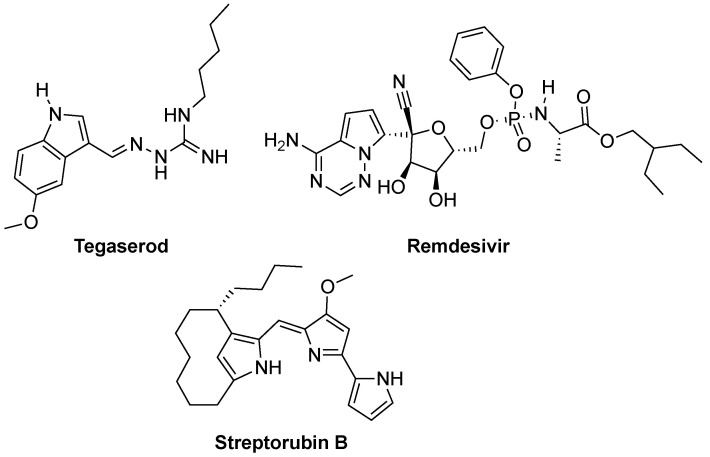 Figure 9
Figure 9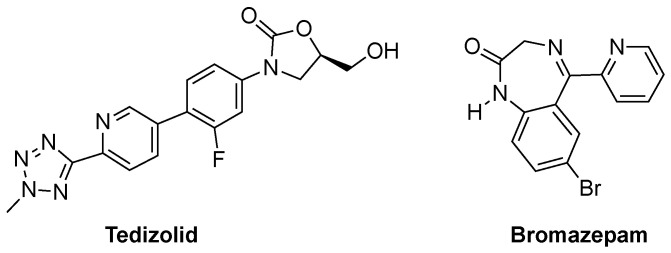 Figure 10
Figure 10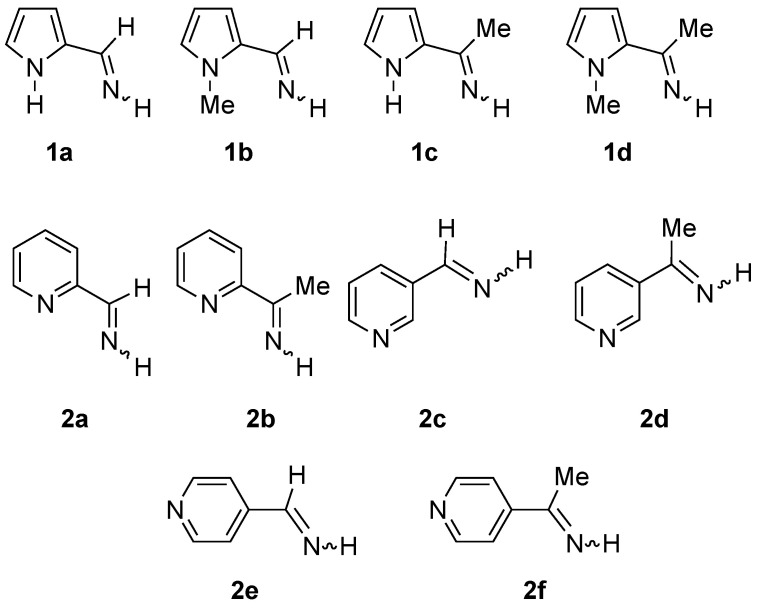 Figure 11
Figure 11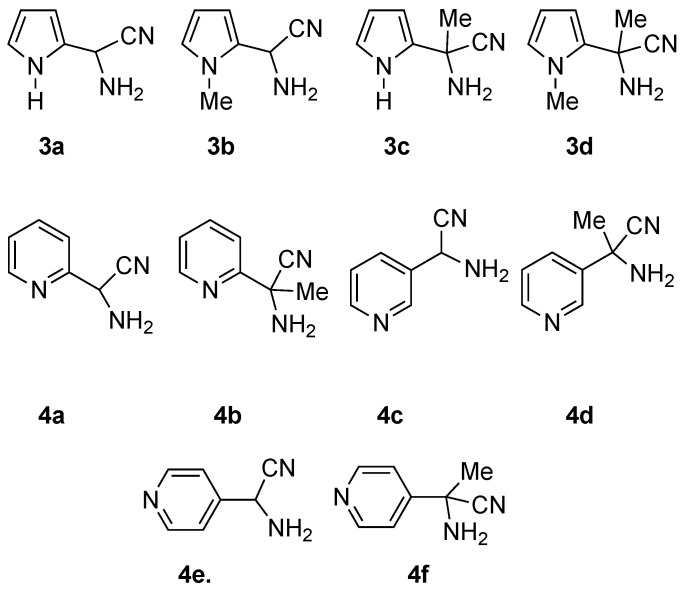 Figure 12
Figure 12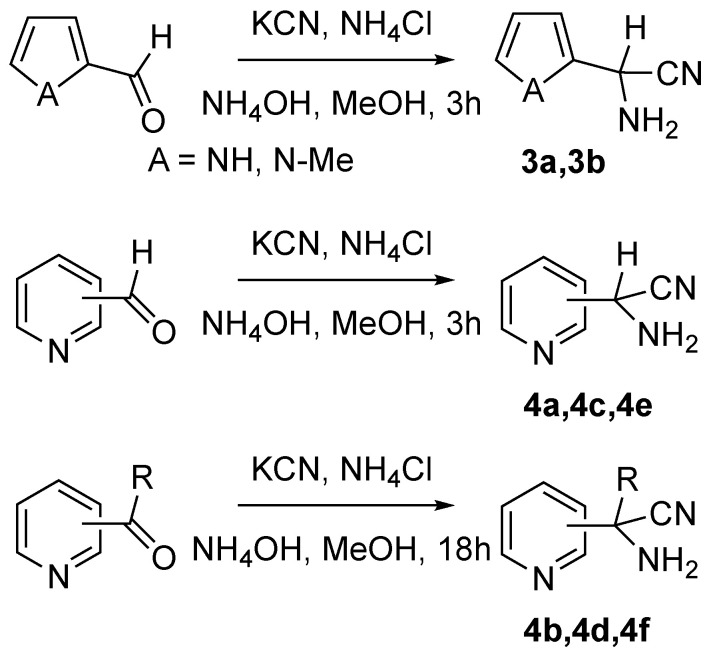 Figure 13
Figure 13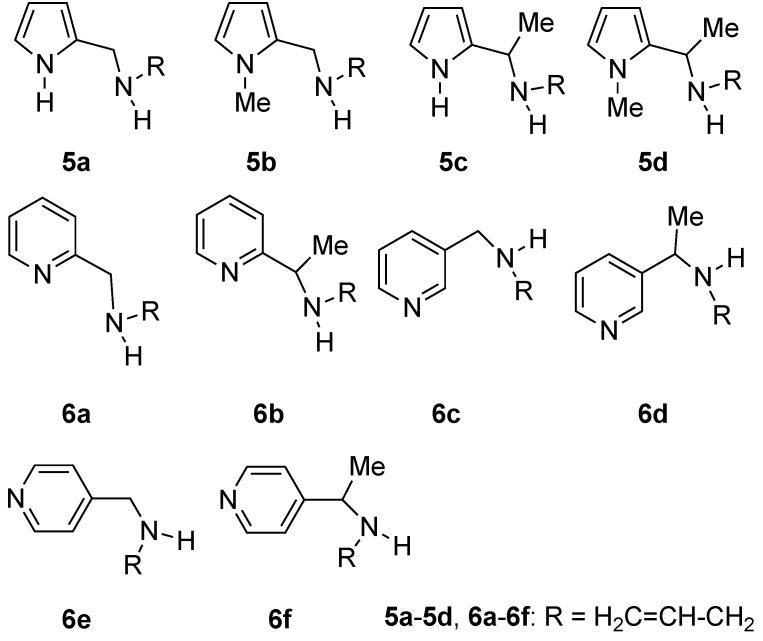 Figure 14
Figure 14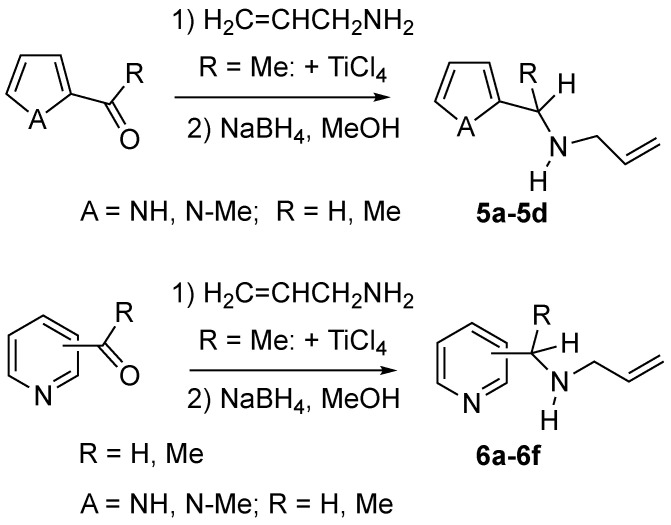 Figure 15
Figure 15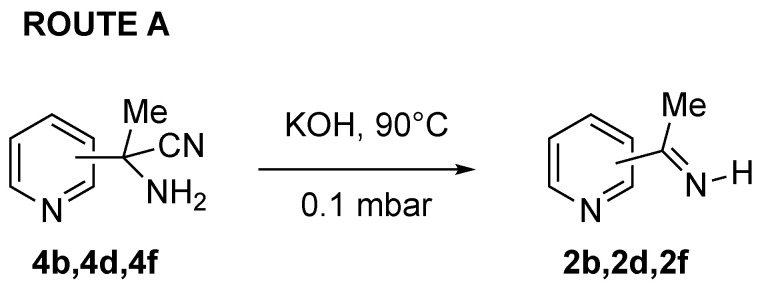 Figure 16
Figure 16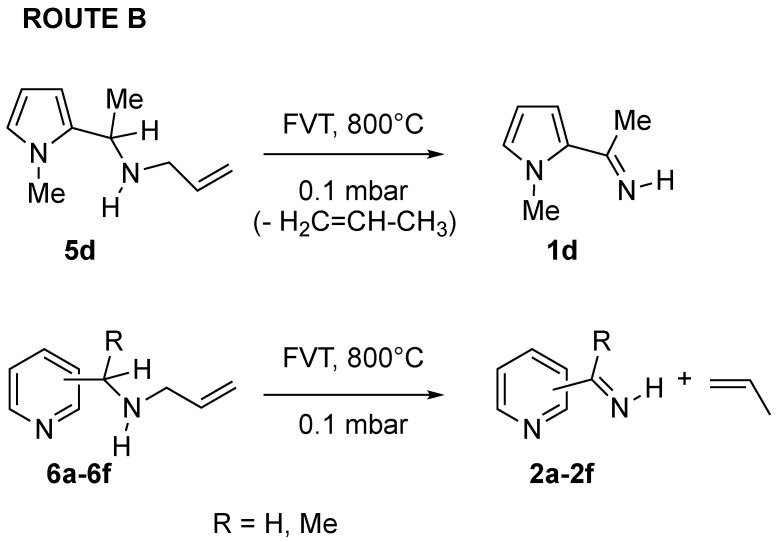 Figure 17
Figure 17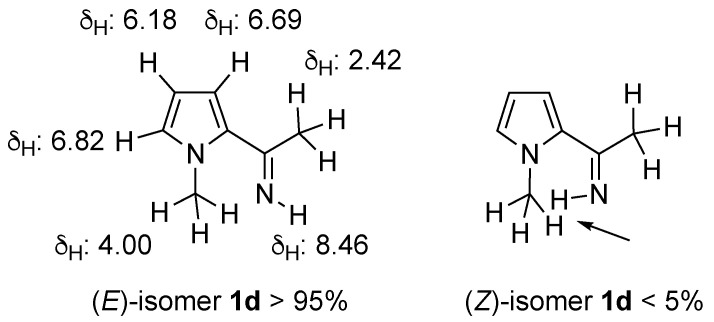 Figure 18
Figure 18 Figure 19
Figure 19 Figure 20
Figure 20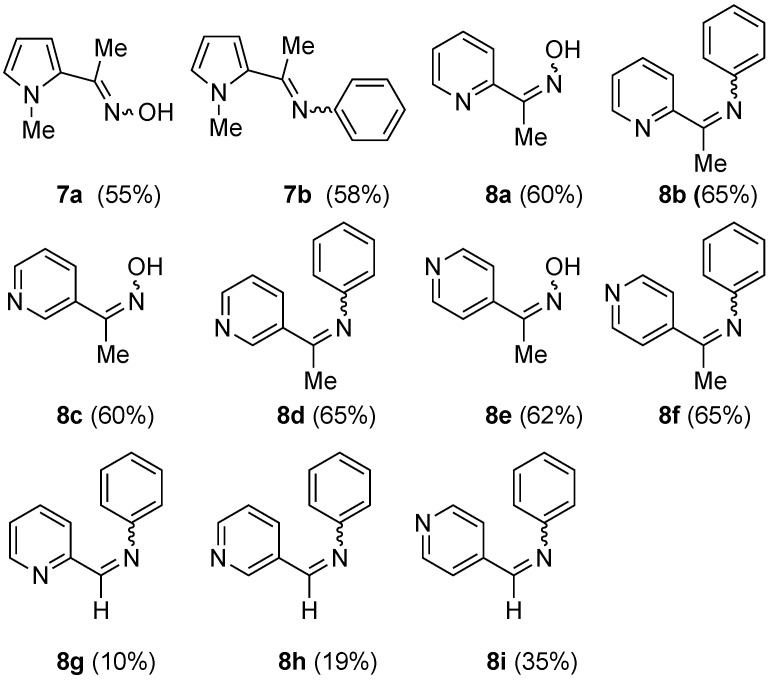 Figure 21
Figure 21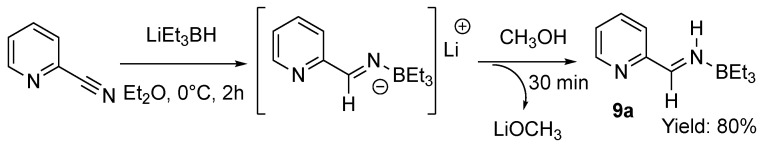 Figure 22
Figure 22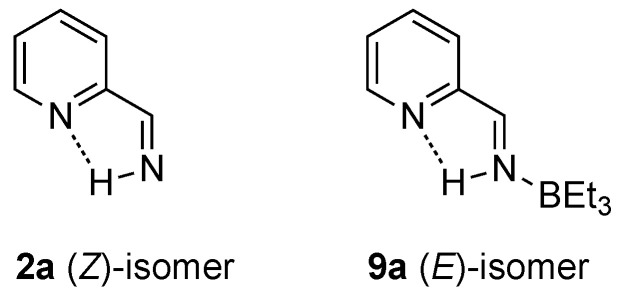 Figure 23
Figure 23Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSynthesis and Reactivity of Heterocycles · Synthesis and Biological Evaluation · Synthesis and Characterization of Pyrroles
1. Introduction
The synthesis of new unstabilized imines has been a challenging subject for many decades. The simplest, CH_2_=NH, was synthesized by a retro-Diels–Alder reaction from an aza-norbornene derivative [1], the dehydrochlorination of N-chloromethylamine [1,2], thermolysis [3], or the photolysis of methyl azide [4] and later by the dehydrocyanation of aminoacetonitrile [5]. Such reactions have been extended to the synthesis of C-monosubstituted derivatives, the N-unsubstituted aldimines, but many with an alkyl substituent [2,5]. On the other hand, the retroenic reaction of secondary allyl- and propargyl-amines provided the expected imine and propene or allene, respectively, opening the way to vinyl- or ethynylimine [6,7]. Thus, very recently, the thermolysis of dipropargylamine followed by the recording of the millimeter wave spectrum of the ethynylimine formed allowed its detection in the interstellar medium (ISM) [7] where methanimine [8], ethanimine [9], cyanomethanimine [10,11], and N-cyanomethanimine [12] have also been observed. On the other hand, the photolysis of an azide deposited on a cold window allowed the first detection by infrared spectroscopy of a functionalized N-H-aldimine, HCOCH=NH [13], or of a diamine, 1,4-diazabutadiene, HN=CH-CH=NH [14]. In a recent article on the theoretical chemistry of candidates for the ISM, it was interesting to see the large number of imines of formula CmHnN_2_ that have never been synthesized [15].
In our approach, where the isolation and chemistry of the synthesized compounds are essential parameters, the preparation of the simplest derivatives of a family of compounds associated with a possible selective addition of substituent(s) allows us to obtain good knowledge of these target species. We have recently reported the syntheses of two imines, HCN dimers, methylenecyanamide (CH_2_=NCN) and (Z)- and (E)-Iminoacetonitriles (NC-CH=NH) [16], and of the derivatives of two new families of N-H aldimines, one substituted by a furan group [17] and the second by a thiophene group [18]. All these compounds were obtained in gas phase, paving the way for their characterization by many techniques (millimetric, infrared or UV-VIS in the gas phase, photoelectron spectroscopy, gas electron diffraction, mass spectrometry). For all synthesized derivatives bearing a heterocyclopentadiene substituent, and even for the most reactive unsubstituted derivatives, transimination reactions were efficiently carried out showing the potential of such compounds as precursors of many imines. However, compared to simple N-H aldimines, we observed an increase in the difficulty of the gas phase transfer of these imines after the reactor or the furnace. We have drawn an analogy with the atmospheric pressure boiling point of furan (31.5 °C) and thiophene (84 °C) and the synthesized imines that are really different, e.g., from the highly volatile C3 imines with the boiling points of ethane (−88.6 °C), ethene (−103.9 °C), or ethyne (−84.7 °C) for comparison. In the literature, the most studied heterocyclopentadienes are unambiguously the furan, thiophene, and pyrrole derivatives. Thus, this study focused on the synthesis, isolation, and chemistry of the third one leading to the pyrrolimines. For pyrrole, a boiling point of 129.7 °C has been reported indicating a probably high difficulty in obtaining pyrrolimines in the gas phase. Due to the trivalent nitrogen atom, a six-membered aromatic ring, pyridine is also a possible substituent. So, we also studied pyridinimines, a family of compounds with a pyridine ring as an aromatic substituent.
Pyrrolimines, as well as furanimines [17] and thiophenimines [18], are of great interest as bioactive and pharmaceutical compounds. As with drugs containing the thiophenimine moiety, the pyrrolimine moiety is often found in polycyclic systems where the C=N group is part of a pyrrole, pyrimidine, or pyridazine ring. For example, Remdesivir [19], Baricitinib [20], prodiginines [21] (prodigiosin R1, metacycloprodigiosin, undecylprodigiosin, roseophilin, streptorubin B) are therefore commercial drugs formally containing pyrrolimine moieties (Scheme 1). Remdesivir, marketed as Veklury, is a broad-spectrum antiviral agent. It has demonstrated efficacy against a broad range of viruses [22], including filoviruses such as Ebola and Marburg, and coronaviruses such as SARS-CoV, MERS-CoV, and SARS-CoV-2 [23,24]. In addition, it was the first antiviral to be shown to be effective and approved for the treatment of coronavirus disease 2019 (COVID-19) [25,26,27]. Tegaserod, on the other hand, has an acyclic imine motif associated with pyrrole. It is a drug used for the treatment of irritable bowel syndrome (IBS) [28,29].
Similarly, pyridinimines are very important motifs in several drugs. They are present, for example, in Tedizolid [30], an antibiotic of the oxazolidinone class, and in Bromazepam, which is an anti-anxiety agent (Scheme 2) [31].
2. Results
2.1. N-H-Unsubstituted Pyrrolimines and Pyridinimines
2.1.1. Selected Imines
Considering only N-unsubstituted imines directly linked to the aromatic ring, we failed in our attempts to find in the scientific literature such compounds with a pyrrole substituent for aldimines as well as for ketimines, including N-alkylpyrrole derivatives; however, as early as 1964, the synthesis of C-alkyl derivatives with a pyridine substituent were reported, and among them were α-methyl-2-, 3-, and 4-pyridinylmethanimines, but without spectroscopic characterization [32]. More recently, the ^1^H and ^13^C NMR spectra and HRMS of the α-methyl-3-pyridinemethanimine have been reported [33]. Pyridinylaldimines were also postulated as intermediates [34].
We selected 10 imines 1a–1d and 2a–2f (Scheme 3). We then considered their synthesis by dehydrocyanation of the corresponding α-aminonitriles and by a retro-ene reaction from N-allylic derivatives. Both precursors will be presented below.
2.1.2. Synthesis of the α-Aminonitriles 3a–3d and 4a–4f
We thus first investigated the synthesis of the ten α-aminonitriles 3a–3d and 4a–4f, potential precursors of these imines (Scheme 4).
The synthesis of compounds 3b–3d has never been reported, while a patent [35] described compound 3a. The α-aminonitriles 3a and 3b were easily synthesized in a Strecker reaction, but their purification was problematic. Distillation in vacuo as well as chromatography on silica gel failed to increase the purity. Attempts to synthesize the two methyl-derivatives 3c and 3d were unsuccessful even when the reaction was carried out for a longer time. These results show a significant difference with the derivatives where the aromatic ring is a furan or a thiophene [17,18]. This could be attributed to the presence of the second nitrogen in the molecule (that of the pyrrole substituent) which could interact with the imine intermediate.
The pyrrole ring is π-excessive and the N electron lone pair is part of the aromaticity, while the pyridine ring is π-deficient or not and its N electron lone pair can interact with electrophiles. Different behaviors are therefore possible for each family of compounds.
Thus, the synthesis of α-aminonitriles 4a–4e have been reported [36,37,38,39,40], and compounds 4a–4f were readily synthesized in a Strecker reaction, although the methyl derivatives required a longer reaction time. The new compound 4f, obtained in a 90% yield, was easily characterized by ^1^H and ^13^C NMR spectroscopy, infrared spectroscopy and mass spectrometry, and a comparison of its NMR spectra with those of the non-methylated compound 4e (Scheme 5, Table 1).
2.1.3. Synthesis of the N-Allylamines 5a–5d and 6a–6f
Similarly, the ten N-allylamines 5a–5d and 6a–6f are potential precursors of these imines 1a–d and 2a–f (Scheme 6).
For both families of compounds, the synthesis of the N-allylamines 5a, 5b, 6a, 6c, and 6e [41,42,43,44,45] has already been reported, but not that of the C-methylated derivatives 5c, 5d, 6b, 6d, and 6f. We synthesized all of them by the addition of allylamine to the corresponding aldehyde or ketone, followed by the reduction with NaBH_4_ of the formed imines to the expected amines [44,46,47]. The N-allylamines were obtained in good yields (89–93%) and characterized by ^1^H and ^13^C NMR spectroscopy and a comparison of their spectra with those reported in the literature for the known compounds. For the others, infrared spectra and high-resolution mass spectra (HRMS) were added to confirm their structure (see Supplementary Materials) (Scheme 7, Table 2).
2.1.4. Synthesis of and Attempts to Synthesize the Imines 1a–1d and 2a–2f
The vaporization of α-aminonitriles on hot powdered potassium hydroxide (90 °C) (route A) and retro-ene reactions by thermolysis at 800 °C of N-allylic derivatives (route B), both under vacuum, were therefore used with the aim of synthesizing imines 1a–1d and 2a–2f.
The problem of the vaporization of α-aminonitrile precursors encountered during the synthesis of furan and mainly thiophene derivatives becomes more important for α-aminonitriles substituted by a pyrrole or pyridine group. We considered this difficulty logical knowing the boiling points of pyridine, pyrrole, and 1-methylpyrrole, which are, respectively, 115, 129, and 112 °C higher than that of furane (31 °C) and thiophene (84 °C), as already reported above. It should also be remembered that compounds with a hydrogen on the nitrogen of the pyrrole are much more reactive than the others, with this hydrogen being able to lead to undesirable reactions.
From the α-aminonitriles 4b, 4d, and 4f, the pyridinimines 2b, 2d, and 2f were obtained in good yields of 75, 72, and 70%, respectively, and with a good purity when the crude product was condensed in a cold trap cooled at −50 °C before revaporization. Similar experiments with α-aminonitriles 3a, 3b, 4a, 4c, and 4e (2.0 mmol) were unsuccessful (Scheme 8, Table 3). The attempt to synthesize 1c and 1d by this route was not carried out because the precursors 3c and 3d were not prepared.
Overall, the synthesis of pyrrolaldimines and pyridinaldimines by the dehydrocyanation reaction is not satisfactory. Only ketimines with a pyridine ring as a substituent have been obtained in good yields and purity. A much higher instability of aldimines—compounds generally more kinetically unstable than the corresponding ketimines—on hot powdered KOH is probably a parameter of these reactions, but a higher instability on heating of the precursors of aldimines than those of ketimines can also be considered. Therefore, in order to have more efficient syntheses, we proceeded to their preparation by route B, a retro-ene reaction from N-allylamines 5a–5d and 6a–6f by thermolysis at 800 °C under flash vacuum thermolysis conditions.
By FVT, N-methyl-α-methyl-2-pyrrolemethanimine 1d and the three pyridineketimines 2b, 2d, and 2f were synthesized with good yields ranging from 77 to 80%. Moreover, this approach also allows the synthesis of the first pyridinaldimines: pyridinemethanimines 2c and 2e were obtained with yields of 76 and 80% in the presence of propene, and characterized by ^1^H and ^13^C NMR spectroscopy. Imine 2a was obtained with a yield of 23% in the presence of impurities whose major products are pyridine and propene. The purification of 2a, 2c and 2e by condensation–revaporization was not successful because they decomposed at a temperature below the vaporization temperature under vacuum (0.1 mbar). On the other hand, the by-products are too abundant with, at best, traces of pyrrolimines 1a–1c to consider that they have been synthesized (Scheme 9, Table 4).
The half-life of pyrrolimine 1d (5% in CDCl_3_ and under dry nitrogen) is about 2 days at room temperature, and no decomposition was observed after 2 days for ketimines 2b, 2d, and 2f in such a dilution. Under the same conditions, the half-life of aldimines 2a, 2c, and 2e was about 10 min. Unidentified decomposition products containing pyridine rings were obtained.
The ^1^H NMR spectrum of imine 1d shows a 19:1 ratio between the two stereoisomers. By NOESY analysis, the main stereoisomer was assigned to the (E) isomer, an expected result given the steric hindrance in the (Z) isomer (see Supplementary Materials and Scheme 10). The N-H signal of (E)-1d and those corresponding to the hydrogens of the pyrrole ring were observed at δ 8.46 ppm and between 6 and 7 ppm, respectively, and the hydrogens of the methyl groups at δ 4.00 and 2.42 ppm. In ^13^C NMR spectroscopy, the carbon of the imine was observed at δ 169.7 ppm, the ring carbons between 105 and 130 ppm, and the methyl groups at δ 39.0 and 29.0 ppm.
The (E)- and (Z)-pyridinaldimines 2a, 2c, and 2e were characterized by NMR spectroscopy: on the ^1^H NMR spectrum, the signal of the proton on the nitrogen is observed around 9.5 ppm except for the (Z)-isomer of 2a at 11.35 ppm; that on the carbon appears around 8.5 ppm. Coupling constants of about 25 and 16 Hz were easily attributed to the (Z)- and (E)-isomer, respectively. The chemical shifts in imines on the ^13^C NMR spectra appear between 167 and 169 ppm. For, the cycle, the ^1^H and ^13^C chemical shifts are quite similar to those of substituted aldimines [43,44,45] (Scheme 11).
2.2. Reactions of Transimination
Since pyrrolimine 1d and pyridine-ketimines 2b, 2d, and 2f are relatively stable kinetically, transimination should proceed readily unless the ring nitrogen interferes in the reaction. In a two-step reaction from N-allylamines, we reacted hydroxylamine and aniline with pyrrole-ketimine 1d and the three pyridine-ketimines (2b, 2d, 2f) to form room-temperature stable N-substituted imines 7a, 7b, and 8a–8f (Scheme 12). The aromatic ring nitrogen does not appear to play any role in this reaction. Eight compounds were thus obtained with good yields ranging from 55 to 65% from the amine (Scheme 13).
Pyridinimines 2c and 2e with propene and the crude mixture containing imine 2a were involved in a transamination reaction at the 0.5 mmol scale with aniline as amine. N-phenylimines 8g–8i were obtained in overall yields of 10 (2a), 19 (2c), and 35% (2e) from amines 6a, 6c, and 6e.
All these N-substituted imines were analyzed by ^1^H and ^13^C NMR spectroscopy, and the results are in agreement with those reported in the literature [47,48,49,50,51,52,53,54,55] and also, for 8g–8i, by comparison with authentic samples [54].
2.3. Synthesis of Complexed Imines
The complexation of imines by the reaction of an imine with triethylborane has never been described, but N-H complexed aldimines can be obtained by reaction of the corresponding nitrile with superhydride followed by addition of methanol [56]. This approach facilitated an efficient synthesis of complexed aryl [56], furan- [17], and thiophenimines [18] but proved much less efficient with alkylnitriles [56]. In the case of complexed N-H pyrrolaldimines and pyridinaldimines, it was interesting to know the role played by the aromatic ring and in particular by the nitrogen atom during such a synthesis.
Our attempts to synthesize a complexed pyrrolimine from 2-cyanopyrrole were unsuccessful even using an excess of superhydride. However, this approach was found to be very efficient to prepare the 2-pyridinimine-triethylborane complex 9a from pyridine-2-carbonitrile (Scheme 14). This complex was obtained in 80% yield and characterized by ^1^H, ^13^C, and ^11^B NMR spectroscopy. Surprisingly, the same reaction with pyridine-3-carbonitrile and pyridine-4-carbonitrile gave a complex mixture of products. Unambiguously, the presence of the nitrogen atom on the pyrrole and pyridine ring considerably influences the reactivity of the corresponding nitriles with superhydride while such reactions were really easy with aryl, furan, or thiophene derivatives.
Only the (E) isomer was observed by ^1^H NMR spectroscopy for compound 9a. The signal of the proton carried by the nitrogen atom is observed as a broad doublet at 10.9 ppm with coupling constants ^3^J = 20.7 Hz. The proton carried by the imine carbon is observed as a doublet at 8.77 ppm.
3. Discussion
Pyrrolimine 1d is a ketimine and an N-methylpyrrole. To our knowledge, compound 1d is the first N-H pyrrolimine characterized and isolated to date. The synthesis of N-H pyrrolaldimines, and N-H pyrrolketimines N-unsubstituted on the nitrogen of the pyrrole ring therefore remains a challenge that will probably require an approach other than the dehydrocyanation or retro-ene reactions of the corresponding precursors. Compound 1d was characterized by ^1^H and ^13^C NMR spectroscopy. It is observed mainly as the (E) isomer, with steric hindrance appearing evident for the (Z) isomer between the hydrogen of the N-H and the methyl group on the other nitrogen (Scheme 10). Compared to the corresponding α-methyl-2-furanmethanimine [17] and α-methyl-2-thiophenemethanimine [18], we observe a chemical shift upfield (δ 8.76 ppm) for the proton signal on the nitrogen atom that could be attributed to a donor effect of the methyl group of the pyrrole moiety. The ^13^C chemical shift in the imine carbon is similar for these three compounds, which otherwise exhibit comparable kinetic stability.
The dehydrocyanation of α-aminonitriles over hot KOH (90 °C) and under vacuum as well as the vacuum thermolysis of N-allylamines provide two routes to synthesize the three N-H pyridineketimines 2b, 2d, and 2f and obtain them in pure form by condensation and revaporization. In addition, this second approach via a retro-ene reaction allows the synthesis of the first pyridinaldimines, the 3-pyridinemethanimine 2c and 4-pyridinemethanimine 2e, which were thus obtained with a yield of 76 and 80%, respectively, in the presence of slightly more than one equivalent of propene, while 2-pyridinemethanimine 2a was obtained in the presence of several impurities and with a lower yield (23%).
These (Z)-isomers of 2-pyridinimines 2a and 2b exhibit a peculiar property with a chemical shift at downfield for the hydrogen linked to the nitrogen. This could be attributed to a hydrogen bond with the nitrogen atom of the cycle. Such a bond has already been proposed for 2-furanemethanimine [17] but not for 2-thiophenemethanimine [18]. This leads, for 2a–2f, to huge differences in the Z/E ratio, with the (E) isomer being more abundant for the 3-pyridine and 4-pyridine derivatives (2c: Z/E = 1/8; 2e: Z/E = 1/17) and not for the 2-pyridine derivative (2a: Z/E = 5/3). Similarly, the usual chemical shift upfield due to complexation [17,18,55,57] was not observed for the (E)-2-pyridinemethanimine-triethylborane complex 9a and could also be attributed to hydrogen bonding (Scheme 15).
Such free pyridinaldimines are kinetically unstable with a very short half-life evaluated to 10 min at room temperature under nitrogen and diluted in a solvent (5% in CD_2_Cl_2_, CDCl_3_, CH_3_CN), showing a higher reactivity than the corresponding furan-, thiophen- and phenyl-aldimines.
The preparation of N-H pyrrolaldimines and pyridinaldimines is therefore much more difficult than for the corresponding furan-, thiophen-, and arylaldimines [17,18,33]. We are clearly, and even more than for thiophenimines, at the limit of an approach to generating the products in the gas phase before condensation on a cold finger. Based on numerous scientific publications on the chemistry of arylaldimines, the chemistry of these pyridinaldimines is currently in progress in our laboratory as well as the search for an approach to synthesize pyrrolaldimines. It should be noted that N-H cyclopentadienimines are also still unknown to date.
4. Materials and Methods
I. Route A: General procedure for the synthesis of imines 2b, 2d, and 2f by dehydrocyanation**.**
A reactor (ϕ = 2.0 cm, L = 40 cm) half-filled with KOH powder (39 g, 0.7 mol) was placed in a vacuum line (0.1 mbar) between a reagent inlet on one side and a solvent inlet, a nitrogen gas inlet, and a cold finger on the other. The KOH was heated to 90 °C by a circulating bath, and α-aminonitriles 4b, 4d, and 4f (2.0 mmol) was slowly vaporized into the reactor. The imine formed was condensed in a U-tube immersed in a cold bath cooled to −50 °C to remove propene. At the end of the reaction, the U-tube was allowed to warm to room temperature, and the imine was condensed on the cold finger cooled with liquid nitrogen. A solvent was added at this step. At the end of the addition, the pump was disconnected, the assembly was filled with dry nitrogen, and the liquid nitrogen in the cold finger was expelled with compressed air. The imine and solvent flowed rapidly upon fusion in the NMR tube or in the flask fitted to the bottom of the cold finger and immersed in a liquid nitrogen bath. Pyridinimines 2b, 2d, and 2f were obtained in good purity and with a good yield by this approach. Similar experiments with α-aminonitriles 3a, 3b, 4a, 4c, and 4e (2.0 mmol), even without intermediate trapping in the U-tube, failed.
II. Route B: General procedure for the synthesis of imines 1d and 2a–2f by retro-ene reaction.
Compounds 6a, 6c, and 6e (0.5 mmol) were vaporized under vacuum (0.1 mbar) in a quartz tube (L = 35 cm, ϕ = 25 mm) heated in an oven to 800 °C, and the products were directly condensed on a cold finger cooled with liquid nitrogen. A solvent was added at this step. It should be noted that heating to about 50 °C with a heat gun of the connection between the oven and the cold finger is necessary to avoid condensation and decomposition of imines 2a, 2c, and 2e on the wall. A solvent was added, and the next step was identical to that reported above. Pyridinimines 2c and 2e were obtained in the presence of slightly more than one equivalent of propene. Several impurities (mainly pyridine and propene) were present with imine 2a. Yields were determined by ^1^H NMR spectroscopy with an internal reference.
Compounds 5d, 6b, 6d, and 6f (2.0 mmol) were vaporized under vacuum in the same way and the products were trapped in a U-tube immersed in a cold bath cooled at −50 °C to remove propene. The next step was identical to the one reported above. Imines 1d, 2b, 2d, and 2f were obtained in good purity and with a good yield by this approach.
Vaporizing 5a–5c with the same approach led to a product mixture too complex to consider that imines 1a–1c were synthesized.
III. Spectroscopic data of imines 1d and 2a–2f. (Mass spectrometry (HRMS) was not performed for known compounds 2b, 2d, and 2e and was not obtained for kinetically unstable compounds 2a, 2c, and 2e.)
N-Methyl-α-methyl-2-pyrrolemethanimine (1d). Yield: 80% (195 mg, 1.6 mmol, route B). E/Z = 19/1. (E) ^1^H NMR (400 MHz, CDCl_3_) δ 8.76 (s, 1H, NH), 6.82 (m, 1H), 6.69 (m, 1H), 6.18 (m, 1H), 4.00 (s, 3H, N-CH_3_), 2.42 (s, 3H, CH_3_). ^13^C{^1^H} NMR (100 MHz, CDCl_3_) δ 168.2, 130.4, 128.9, 115.5, 107.1, 38.9, 29.1. IR (KBr, film, cm^−1^): 3333 (m), 1609 (m, ν_C=N_), 1432 (s), 1289 (m). HRMS (ASAP): [M + H]^+^ calculated for C_7_H_11_N_2_^+^ m/z 123.09167, found m/z 123.0916.
2-Pyridinemethanimine (2a). Yield: 23% (0.12 mmol, route B). (τ_1/2_ (5% (crude) in CDCl_3_ ≈ 7 min). Z/E = 5/3. (Z) ^1^H NMR (400 MHz, CD_2_Cl_2_, 203 K) δ 11.35 (d, ^3^J = 24.4 Hz, 1H, NH), 8.66 (m, 1H), 8.46 (d, ^3^J = 24.4 Hz, 1H, CH=N), 7.85 (m, 1H) 7.78 (m, 1H), 7.38 (m, 2H). ^13^C{^1^H} NMR (100 MHz, CD_2_Cl_2_, 203 K) δ 167.2, 155.1, 149.8, 138.2, 126.4, 121.6 (E) ^1^H NMR (400 MHz, CD_2_Cl_2_, 203 K) δ 10.53 (d, ^3^J = 16.5 Hz, 1H, NH), 8.75 (d, ^3^J = 16.5 Hz, 1H, CH=N), 8.60 (m, 1H), 8.10 (m, 1H), 7.5–7.4 (m, 2H). ^13^C{^1^H} NMR (100 MHz, CD_2_Cl_2_, 203 K) δ 171.6, 155.1, 149.6, 137.9, 124.8, 120.0.
α-Methyl-2-pyridinemethanimine (2b) [32]. Yield: 75% (180 mg, 1.50 mmol, route A), 80% (192 mg, 1.60 mmol, route B). Z/E = 3/1. (Z)^1^H NMR (400 MHz, CD_2_Cl_2_, 203 K) δ 10.98 (s, 1H, NH), 8.42 (m, 1H), 7.60 (m, 1H), 7.40 (m, 1H), 7.14 (m, 1H), 2.29 (s, 3H, CH_3_). ^13^C{^1^H} NMR (100 MHz, CD_2_Cl_2_, 203 K) δ 171.7, 150.1, 149.3, 137.7, 125.1, 120.8, 22.2. (E) ^1^H NMR (400 MHz, CD_2_Cl_2_, 203 K) δ 9.62 (s, 1H, NH), 8.42 (m, 1H), 8.00 (m, 1H), 7.57 (m, 1H), 7.14 (m, 1H), 2.50 (s, 3H, CH_3_). ^13^C{^1^H} NMR (100 Hz, CD_2_Cl_2_, 203 K) δ 175.9, 155.5, 148.4, 136.6, 125.1, 121.0, 26.0. IR (KBr, film, ν cm^−1^): 3396 (s), 1645 (m, ν_C=N_), 1459 (m).
3-Pyridinemethanimine (2c). Yield: 76% (40 mg, 0.38 mmol, route B). (τ_1/2_ (5% in CDCl_3_ ≈ 10 min). E/Z = 8/1. (E) ^1^H NMR (400 MHz, CD_2_Cl_2_, 198 K) δ 10.23 (d, ^3^J = 16.3 Hz, 1H, NH), 8.87 (s, 1H), 8.77 (d, 1H, ^3^J = 16.3 Hz, CH=N), 8.67 (s, 1H), 8.25 (m, 1H), 7.45 (m, 1H). ^13^C{^1^H} NMR (100 MHz, CD_2_Cl_2_, 198 K) δ 168.1, 152.4, 150.6, 134.2, 132.0, 124.2. (Z) ^1^H NMR (400 MHz, CD_2_Cl_2_, 198 K) δ 10.52 (d, ^3^J = 25.2 Hz, 1H, NH), 8.88 (s, 1H), 8.78 (d, ^3^J = 25 Hz, 1H, CH=N); the 3 other signals were not unambiguously identified.
α-Methyl-3-pyridinemethanimine (2d) [32,33]. Yield: 72% (173 mg, 1.44 mmol, route A), 77% (185 mg, 1.54 mmol, route B). E/Z = 4/1. (E)^1^H NMR (400 MHz, CDCl_3_, 223 K) δ 9.54 (s, 1H, NH), 9.06 (m, 1H), 8.70 (m, 1H), 8.30 (m, 1H), 7.42 (m, 1H), 2.53 (s, 3H, CH_3_). ^13^C{^1^H} NMR (100 MHz, CDCl_3_, 223 K) δ 173.1, 151.8, 148.5, 134.6, 133.4, 123.8, 27.4. (Z) ^1^H NMR (400 MHz, CDCl_3_, 223 K) δ 9.80 (s, 1H, NH), 8.82 (m, 1H), 8.61 (m, 1H), 7.89 (m, 1H), 7.28 (m, 1H), 2.53 (s, 3H, CH_3_). IR (KBr, film, ν cm^−1^): 3411 (s), 1625 (m, ν_C=N_), 1409 (f).
4-Pyridinemethanimine (2e). Yield: 80% (42 mg, 0.4 mmol, route B). (τ_1/2_ (5% in CDCl_3_ ≈ 10 min). E/Z = 17/1. (E)^1^H NMR (400 MHz, CD_2_Cl_2_, 198 K) δ 10.52 (d, ^3^J = 16.1 Hz, 1H, NH), 8.71 (m, 2H), 8.70 (d, ^3^J = 16.1 Hz, 1H, CH=N), 7.68 (m, 2H). ^13^C{^1^H} NMR (100 Hz, CD_2_Cl_2_, 198 K) δ 169.0, 150.7, 143.0, 121.8. (Z) ^1^H NMR (400 MHz, CD_2_Cl_2_, 198 K) δ 10.95 (d, ^3^J = 25.4 Hz, 1H, NH), 8.80 (d, ^3^J = 25.4 Hz, 1H, CH=N), 8.48 (m, 2H), 7.48 (m, 2H).
α-Methyl-4-pyridinemethanimine (2f) [32]. Yield: 70% (168 mg, 1.40 mmol, route A), 80% (192 mg, 1.60 mmol, route B). E/Z = 4/1. (E)^1^H NMR (400 MHz, CDCl_3_, 223 K) δ 9.78 (s, 1H, NH), 8.65 (m, 2H), 7.70 (m, 2H), 2.43 (s, 3H, CH_3_). ^13^C{^1^H} NMR (100 Hz, CDCl_3_, 223 K) δ 173.6, 150.4, 144.9, 121.2, 27.0. (Z) ^1^H NMR (400 MHz, CDCl_3_, 223 K) δ 9.97 (s, 1H, NH), 8.60 (m, 2H), 7.38 (m, 2H), 2.43 (s, 3H, CH_3_). ^13^C{^1^H} NMR (100 MHz, CDCl_3_, 223 K) δ 175.1, 150.7, 144.9, 119.9, 24.3. IR (KBr, film, ν cm^−1^): 3440 (s), 1643 (m, ν_C=N_), 1404 (f).
IV. Synthesis of pyridinemethanimine-triethylborane complex (9a). To a solution of pyridine-2-carbonitrile (0.505 g, 4.85 mmol) in diethyl ether (10 mL), lithium triethylborohydride (1M in THF; 4.85 mL, 4.85 mmol) was added at 0 °C, and the solution was stirred for 2 h at 0 °C. Methanol (155 mg, 4.85 mmol) was added to the reaction mixture and stirred for 30 min. The solvent was then evaporated under reduced pressure and the residue was dissolved in dry pentane (5 mL) and filtered through a Kramer filter. Pyridinemethanimine-triethylborane complex 9a was then precipitated out by slowly cooling the solution to −80 °C, removing the liquid using a pipette, and drying in vacuo. Only the (E) isomer of 9a was obtained in good yield (80%). It is stable at room temperature. (Mass spectrometry (HRMS) analyses are never obtained for such compounds [17,18,55,57].)
(E)-2-pyridinemethanimine-triethylborane complex (9a). Yield: 80% (0.79 g, 3.9 mmol). ^1^H NMR (400 MHz, CDCl_3_) δ 10.91 (d, ^3^J = 20.7 Hz, 1H, NH), 8.77 (d, ^3^J = 4.8 Hz, 1H, CH=N), 8.11, 7.94, 7.61, 7.55 (m, 4H, H-cycle), 0.76 (t, ^3^J = 7.8 Hz, 9H, 3 CH_3_), 0.36 (d, J = 7.9 Hz, 6H, 3 CH_2_). ^13^C{^1^H} NMR (100 MHz, CDCl_3_) δ 160.7, 150.7, 146.2, 138.0, 127.6, 126.2, 9.7. ^11^B{^1^H} NMR (128 MHz, CDCl_3_) δ -2.20. IR (KBr, film, ν cm^−1^): 3295 (m), 2895 (vs), 1656 (m, ν_C=N_), 1400 (s), 1265 (m).
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Braillon B. Lasne M.-C. Ripoll J.-L. Denis J.M. Methanimine Nouv. J. Chim.19826121122
- 2Guillemin J.-C. Denis J.-M. Vacuum dynamic gas-phase/solid-phase reactions: N-chlorination of primary amines and the α-elimination of the resulting chloramines: Access to reactive (E)- and (Z)-alkanimines Angew. Chem. Int. Ed.19822169010.1002/anie.198206901 · doi ↗
- 3Bock H. Dammel R. Gas-phase reactions. 66. Gas-phase pyrolyses of alkyl azides: Experimental evidence for chemical activation J. Am. Chem. Soc.19881105261526910.1021/ja 00224 a 004 · doi ↗
- 4Ying L. Xia Y. Shang H. Zhao X. Tang Y. Photodissociation of methylazide: Observation of triplet methylnitrene radical J. Chem. Phys.19961055798580510.1063/1.472423 · doi ↗
- 5Guillemin J.-C. Denis J.-M. Synthèse d’imines linéaires non-stabilisées par réactions gaz-solide sous vide (1)Tetrahedron 1988444431444610.1016/S 0040-4020(01)86145-7 · doi ↗
- 6Earl R.A. Vollhardt K.P.C. On the synthetic utility of thermally generated imines: The retro-ene imino Diels-Alder reaction Heterocycles 19821926527110.1002/chin.198232122 · doi ↗
- 7Bizzocchi L. Prudenzano D. Rivilla V.M. Pietropolli-Charmet A. Giuliano B.M. Caselli P. Martín-Pintado J. Jiménez-Serra I. Martín S. Requena-Torres M.A. Propargylimine in the laboratory and in space: Millimetre-wave spectroscopy and first detection in the ISM Astron. Astrophys.2020640 A 9810.1051/0004-6361/202038083 · doi ↗
- 8Godfrey P.D. Brown R.D. Robinson B.J. Sinclair M.W. Discovery of Interstellar Methanimine (Formaldimine)Astrophys. J. Lett.197313119