Therapeutic Challenges and Emerging Strategies for T674I and PTPN11 Mutations in a FIP1L1-PDGFRA-Positive Myeloproliferative Neoplasm: A Case Report
Sıdıka Gülkan Özkan, Ali Kimiaei, Seyedehtina Safaei, Mutlu Karkucak, Mustafa Nuri Yenerel, Aslı Yüksel Öztürkmen, Burak Alp, Hasan Atilla Özkan

TL;DR
This case report describes a rare and aggressive myeloproliferative neoplasm with resistance to standard treatments due to specific mutations.
Contribution
The first reported case of concurrent FIP1L1-PDGFRA T674I and PTPN11 mutations in a myeloproliferative neoplasm.
Findings
The patient initially responded to imatinib but developed resistance after ten months.
Despite multiple treatments, including allo-HSCT, the disease progressed and the patient died from multiorgan failure.
The case highlights the challenges of treating FIP1L1-PDGFRA T674I-positive disease with PTPN11 mutation.
Abstract
Myeloproliferative neoplasm (MPN) with eosinophilia associated with FIP1L1-PDGFRA is a rare eosinophilic disorder typically treated with imatinib. However, resistance due to the T674I mutation poses a significant challenge. This case presents the first reported instance of concurrent FIP1L1-PDGFRA T674I and PTPN11 (p.E76D) mutations in a 38-year-old male patient with MPN and eosinophilia. The patient initially responded to imatinib but developed resistance after ten months, leading to severe spinal cord compression caused by granulocytic sarcoma. Despite undergoing radiotherapy, chemotherapy, and allogeneic hematopoietic stem cell transplantation (allo-HSCT), the disease progressed. Although full donor chimerism was achieved post-transplant, the patient relapsed shortly afterward with eosinophilia, splenomegaly, and constitutional symptoms. Further treatments, including sorafenib and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1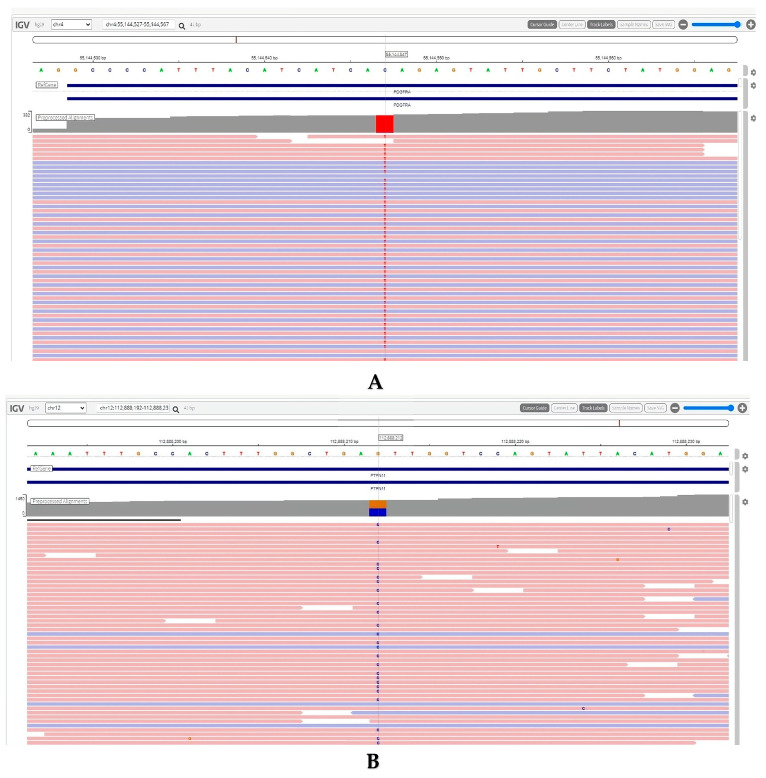 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsEosinophilic Disorders and Syndromes · Interstitial Lung Diseases and Idiopathic Pulmonary Fibrosis · Eosinophilic Esophagitis
1. Introduction
Hypereosinophilic syndrome (HES) is a rare hematologic disorder characterized by the chronic overproduction of eosinophils in the bone marrow, leading to elevated eosinophil counts in the bloodstream [1]. The criteria used to define HES are as follows: (1) blood eosinophilia of ≥1500/mm^3^ lasting for more than 6 months (or death before 6 months with signs and symptoms suggestive of hypereosinophilic disease), (2) absence of evidence indicating parasitic, allergic, or other known causes of eosinophilia, and (3) probable signs of organ involvement, including heart failure, gastrointestinal issues, central nervous system abnormalities, fever, or weight loss [2,3].
If secondary causes of eosinophilia are excluded, the next step is to assess for a primary bone marrow disorder [4]. This involves analyzing the blood smear and conducting tests for circulating blasts, dysplastic cells, monocytosis, and elevated levels of serum B12 or tryptase [4]. These findings, combined with bone marrow analysis through morphological, cytogenetic, and immunophenotypic evaluation, can assist in determining whether the differential diagnosis of eosinophilia includes a clearly defined myeloid neoplasm according to the World Health Organization [4].
Myeloproliferative neoplasm (MPN) with eosinophilia associated with FIP1L1-PDGFRA is a form of hypereosinophilic syndrome [4]. It falls under the category of myeloid/lymphoid neoplasms with eosinophilia and rearrangements involving PDGFRA, PDGFRB, FGFR1, or PCM1-JAK2, as classified in the 2016 World Health Organization classification of myeloid neoplasms [4].
All HESs are expected to have an annual age-adjusted incidence rate of 0.18 to 0.36 per 100,000 person-years [1]. Only a small percentage of affected individuals have genetic abnormalities, with the most prevalent being fusion involving FIP1L1-PDGFRA. According to recent data, the average yearly incidence of FIP1L1-PDGFRA-positive myeloid neoplasm with eosinophilia is 0.18 cases per one million people, with a predominance in males [5].
Imatinib mesylate, a tyrosine kinase inhibitor, is the first-line treatment for individuals with the FIP1L1-PDGFRA mutation [6]. The therapeutic effects have been demonstrated with a low dose of 100–200 mg per day, which resulted in an excellent response, while a lower dose of 100–200 mg per week effectively maintained remission [7]. Although rare, resistance to imatinib poses significant treatment challenges and has been predominantly linked to the T674I mutation [7]. In this report, we present a case of a patient with FIP1L1-PDGFRA T674I-positive MPN with eosinophilia, harboring a PTPN11 (p.E76D) mutation. Figure 1 illustrates the therapeutic timeline, genetic mutations, and treatment challenges encountered in this patient.
2. Detailed Case Description
A 38-year-old male patient presented with weakness, bone pain, and B symptoms, prompting him to seek medical attention. He had no known history of chronic illnesses. Physical examination was normal, without massive splenomegaly. Bone marrow examination revealed marked eosinophilia without excess blasts. The patient was diagnosed with FIP1L1-PDGFRA-positive myeloid with eosinophilia. A PET CT performed at diagnosis showed no additional findings of advanced splenomegaly.
After initiation of Imatinib 100 mg/day, symptoms were controlled, and a hematologic response was achieved. Molecular testing was not performed during treatment. Ten months after starting Imatinib, severe bone pain began, followed by sudden weakness in the lower extremities. Spinal MRI revealed a 9 cm mass severely compressing the spinal cord at the C7-T3 vertebrae. Decompression surgery and mass biopsy were performed, with the vertebral mass biopsy reported as granulocytic sarcoma.
A PET CT performed 10 months after the first presentation due to the newly occurring vertebral mass showed supraclavicular and cervical lymph nodes with lower FDG uptake than the liver, right pleural effusion (2.5 cm diameter, SUVmax: 4.2), newly developed mediastinal lymph nodes (SUVmax: 3.1), a hypermetabolic area in the rectum (SUVmax: 7.9), a newly developed C7-D2 vertebral mass (SUVmax: 9.2), and a newly developed vertebral mass in D7 (SUVmax: 4.5). Additionally, multiple hypermetabolic foci were observed on the left humerus head, ribs, and vertebrae. A bone marrow biopsy showed eosinophilic involvement without excess blasts.
Radiotherapy was applied to the involved vertebral column area. Imatinib 100 mg/day was continued, and a 3 + 7 chemotherapy regimen was started after radiotherapy 11 months after the first presentation. Thirteen months after the first presentation, a high-dose ARA-C regimen was initiated. MRI scan showed no significant regression in the mass size after treatment, and B symptoms recurred. The patient was referred to our institution for allogeneic hematopoietic stem cell transplantation (allo-HSCT) due to refractory disease.
On initial physical examination, the patient was conscious and cooperative, with a fever of 38.1 °C. Lung and heart auscultation were normal, with mild tachycardia. Ten-centimeter splenomegaly was noted. Muscle strength of the lower extremities was 2/5.
Initial laboratory values prior to hospitalization were as follows: WBC 37,000/μL, RBC 2.9 million/μL, Hb 8.3 g/dL, MCV 85 fL, RDW 18%, platelets 26,000/μL, neutrophil count 18,600/μL, lymphocyte count 1500/μL, and eosinophil count 16,400/μL. A bone marrow biopsy showed an increase in eosinophilic series, maturation arrest at the myelocyte and metamyelocyte stages, a marked decrease in erythroid series and megakaryocytes, a 1–2/3 reticulin fiber increase, and 5% myeloblasts.
A PET-CT was performed and compared with the pretreatment PET-CT, showing a significant response in lymph nodes and pleural fluid, a complete response in vertebral masses, and a partial response in bone lesions. The FIP1L1-PDGFRA test was positive by FISH analysis. Next-generation sequencing was performed using the Archer FusionPlex Myeloid Panel (originally developed by ArcherDX, headquartered in Boulder, Colorado, USA. In December 2022, ArcherDX was acquired by Integrated DNA Technologies (IDT), based in Coralville, Iowa, USA) (RNA extraction with kit ReliaPrep FFPE (Promega Corporation, Madison, WI, USA) total RNA-Promega, RNA amplification and sequencing with Illumina-Novaseq 6000 (Illumina, Inc., San Diego, CA, USA), data analysis with Archer^®^ Analysis Version: 7.3.2 software) and revealed genomic alterations in the PDGFRA gene (NM_006206.5) at position c.2021C > T (p.Thr674Ile) and in the PTPN11 gene (NM_002834.4) at position c.228G > C (p.Glu76Asp) (Figure 2).
The FLAG-IDA treatment regimen was initiated with sorafenib (off-label use). All symptoms resolved with treatment. However, bone pain, B symptoms, and eosinophilia recurred with hematologic recovery.
Due to refractory disease, after the MEL/FLU/TBI (Melphalan 140 mg/m^2^ on day −11, Fludarabine 100 mg/m^2^ between days −5 and −2, TBI 8 Gy between days −3 and −2) conditioning regimen, allogeneic hematopoietic stem cells were transplanted from an HLA-matched, blood group-compatible sister 16 months after the first presentation. Post-transplant cyclophosphamide and cyclosporine were used for graft-versus-host disease prophylaxis. Neutrophil and platelet engraftment occurred on days +14 and +15, respectively. During the neutropenic period, the patient experienced febrile neutropenia and grade 2–3 mucositis.
Chimerism at day +28 showed 100% donor cells in the peripheral blood. B symptoms resolved, and organomegaly was not found. Sorafenib 400 mg twice daily was continued as a maintenance treatment.
Chimerism at day +56 showed 53% donor cells in peripheral blood. A few days later, B symptoms, splenomegaly, and eosinophilia recurred. On day 60 post-allo-HSCT, Decitabine 20 mg/m^2^ for 5 days and Venetoclax 400 mg/day for 14 days were initiated. Sorafenib was discontinued during this period. On the 15th day of treatment, the patient developed jaundice, pleural, and pericardial effusions, and pericardiocentesis was performed due to cardiac tamponade. Pericardial fluid analysis revealed leukocytosis with increased eosinophils. Donor lymphocyte infusion was administered for the refractory disease. Despite these interventions, on day +85 post-allo-HSCT, the patient died due to multiorgan failure and refractory disease.
3. Discussion
This case highlights the formidable therapeutic challenges of FIP1L1-PDGFRA T674I-positive MPN with eosinophilia with concurrent PTPN11 (p.E76D) mutation. Despite aggressive multimodal treatment including imatinib, sorafenib, chemotherapy, radiotherapy, and allo-HSCT, the patient experienced rapid disease progression and a poor outcome. The coexistence of T674I PDGFRA and PTPN11 mutations likely drove the aggressive clinical course and therapeutic resistance. No standardized treatment protocol exists for FIP1L1-PDGFRA-positive MPN harboring the T674I mutation, particularly with additional genetic alterations. This case emphasizes the urgent need for novel therapeutic approaches targeting complex resistance mechanisms in this rare and challenging patient population.
To our knowledge, this is the first reported case of FIP1L1-PDGFRA-positive MPN with concurrent T674I and PTPN11 (p.E76D) mutations. While the role of PTPN11 mutations in FIP1L1-PDGFRA-positive MPN has not been previously described, it is considered that this mutation may have contributed to the resistance mechanism observed in this patient.
PTPN11 encodes SHP-2, a protein tyrosine phosphatase that plays a critical role in the RAS/MAPK signaling pathway, which regulates cell proliferation, differentiation, and survival [8]. Germline mutations in the PTPN11 gene account for approximately 40% to 50% of Noonan syndrome (NS) cases, a prevalent autosomal dominant disorder marked by facial features, proportionate short stature, and heart defects [9]. Individuals with NS often show signs of myeloproliferative disorders (MPD), typically presenting as transient leukocytosis (elevated white blood cell count) and splenomegaly (enlarged spleen), which typically resolve without long-term effects [9]. In rare instances, however, these patients may develop juvenile myelomonocytic leukemia (JMML) [9].
Somatic gain-of-function mutations in this gene, such as p.Glu76Asp, lead to hyperactivation of this pathway and have been implicated in various hematological malignancies, including JMML, acute myeloid leukemia (AML), B-cell acute lymphoblastic leukemia (B-ALL), and myelodysplastic syndromes (MDS) [8]. In this case, the PTPN11 (p.Glu76Asp) mutation may have contributed to the aggressive disease course and treatment resistance by promoting cell proliferation, survival, and resistance to apoptosis.
In eosinophilic disorders, for M/LN-eo linked to ETV6::ABL1, although available data are limited, mutations in genes such as ARID2, TP53, SETD2, CDKN1B, PTPN11, and SMC1A have been observed in roughly 50% of cases [10]. Likewise, in M/LN-eo with FLT3 fusions, mutations in genes including ASXL1, PTPN11, RUNX1, SETBP1, SRSF2, STAT5B, TET2, TP53, and U2AF1 have been found in approximately 40% to 50% of cases [10].
In the context of FIP1L1-PDGFRA-positive MPN with eosinophilia, the concurrent PTPN11 mutation could have amplified downstream signaling, making the leukemic cells less dependent on PDGFRA-driven pathways and more resistant to tyrosine kinase inhibitors like imatinib. This dual activation of signaling pathways (PDGFRA and RAS/MAPK) may explain the rapid development of resistance and the poor response to subsequent therapies, including sorafenib and decitabine. Furthermore, PTPN11 mutations are known to confer resistance to chemotherapy and targeted therapies in other cancers, which could account for the failure of high-dose ARA-C in this case [11]. Targeting the RAS/MAPK pathway (e.g., with MEK inhibitors) in combination with PDGFRA inhibitors could be a potential strategy to overcome resistance in similar cases, although this remains speculative and requires further investigation [8]. While both PTPN11 mutations and PDGFRA rearrangements independently contribute to hematologic malignancies, there is limited evidence to suggest a direct interaction between these genetic alterations in the pathogenesis of such diseases. Further research is needed to elucidate any potential cooperative roles between PTPN11 mutations and PDGFRA activation in hematologic malignancies. This case is significant as it represents a genetic association that has been described for the first time in the literature.
Treatment options for FIP1L1-PDGFRA-positive MPN with eosinophilia with T674I mutation remain challenging. While imatinib is highly effective for FIP1L1-PDGFRA-positive MPN with eosinophilia, the T674I mutation confers resistance, necessitating alternative approaches. For instance, nilotinib failed to elicit a response in one patient, prompting a switch to sorafenib [12]. Sorafenib has shown in vitro activity against T674I mutants, but clinical responses are often transient due to the emergence of additional resistant mutations like D842V [12,13,14]. Ponatinib, a third-generation tyrosine kinase inhibitor, has demonstrated in vitro efficacy against both T674I and D842V mutations [15]. However, compound mutations can develop, leading to disease progression even under ponatinib treatment [15]. Crenolanib has shown promise in preclinical studies, particularly against T674I mutants, but clinical efficacy remains to be established [15].
The rapid development of resistance to various tyrosine kinase inhibitors highlights the need for novel therapeutic strategies. New biologic treatments, including mepolizumab, reslizumab, and benralizumab, have shown effectiveness in managing hypereosinophilic syndrome (HES), especially in severe or treatment-resistant cases [16]. Mepolizumab and reslizumab reduce eosinophil levels, while benralizumab induces eosinophil death through antibody-dependent cell-mediated cytotoxicity [16]. Mepolizumab is FDA-approved for eosinophilic granulomatosis with polyangiitis (EGPA) and can reduce the need for steroids, although its benefits in steroid-resistant HES are limited [17]. Benralizumab is approved for eosinophilic asthma, and its role in HES is still being investigated [18]. Other promising treatments, such as dexpramipexole, AK002/lirentelimab, dupilumab, and JAK2 inhibitors, are also under investigation for HES management [16].
In cases of refractory disease or blast transformation, -HSCT may be considered as a potentially curative option [19]. A retrospective study conducted by the Chronic Malignancies Working Party of the European Society for Blood and Marrow Transplantation (EBMT) revealed that the preferences for conditioning regimens and T cell depletion strategies were heterogeneous [20]. The reported cohort underwent allo-HSCT over an extended period of time [20]. However, outcomes remain poor.
4. Conclusions
This case highlights the complexities involved in treating FIP1L1-PDGFRA T674I-positive eosinophilic disorder, especially when compounded by additional mutations like PTPN11. Despite initial responses to imatinib and allo-HSCT, the rapid emergence of resistance emphasizes the limitations of current treatment options. These complex resistance patterns call for the development of new targeted therapies and combination strategies to enhance outcomes in such challenging cases. The poor prognosis associated with refractory cases involving multiple mutations further underscores the urgent need for innovative targeted treatments and combination approaches to improve outcomes in this rare and difficult-to-treat condition.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Crane M.M. Chang C.M. Kobayashi M.G. Weller P.F. Incidence of myeloproliferative hypereosinophilic syndrome in the United States and an estimate of all hypereosinophilic syndrome incidence J. Allergy Clin. Immunol.201012617918110.1016/j.jaci.2010.03.03520639012 PMC 5781228 · doi ↗ · pubmed ↗
- 2Chusid M.J. Dale D.C. West B.C. Wolff S.M. The hypereosinophilic syndrome: Analysis of fourteen cases with review of the literature Medicine 1975541271090795 · pubmed ↗
- 3Simon H.-U. Rothenberg M.E. Bochner B.S. Weller P.F. Wardlaw A.J. Wechsler M.E. Rosenwasser L.J. Roufosse F. Gleich G.J. Klion A.D. Refining the definition of hypereosinophilic syndrome J. Allergy Clin. Immunol.2010126454910.1016/j.jaci.2010.03.04220639008 PMC 3400024 · doi ↗ · pubmed ↗
- 4Gotlib J. World Health Organization-defined eosinophilic disorders: 2017 update on diagnosis, risk stratification, and management Am. J. Hematol.2017921243125910.1002/ajh.2488029044676 · doi ↗ · pubmed ↗
- 5Rohmer J. Couteau-Chardon A. Trichereau J. Panel K. Gesquiere C. Ben Abdelali R. Bidet A. BladéJ. Cayuela J. Cony-Makhoul P. Epidemiology, clinical picture and long-term outcomes of FIP 1L 1-PDGFRA-positive myeloid neoplasm with eosinophilia: Data from 151 patients Am. J. Hematol.2020951314132310.1002/ajh.2594532720700 · doi ↗ · pubmed ↗
- 6Klion A. Hypereosinophilic syndrome: Approach to treatment in the era of precision medicine Hematol. Am. Soc. Hematol. Educ. Program 2018201832633110.1182/asheducation-2018.1.32630504328 PMC 6245960 · doi ↗ · pubmed ↗
- 7Qu S.-Q. Qin T.-J. Xu Z.-F. Zhang Y. Ai X.-F. Li B. Zhang H.-L. Fang L.-W. Pan L.-J. Hu N.-B. Long-term outcomes of imatinib in patients with FIP 1L 1/PDGFRA associated chronic eosinophilic leukemia: Experience of a single center in China Oncotarget 20167332293323610.18632/oncotarget.890627120808 PMC 5078089 · doi ↗ · pubmed ↗
- 8Kanumuri R. Kumar Pasupuleti S. Burns S.S. Ramdas B. Kapur R. Targeting SHP 2 phosphatase in hematological malignancies Expert Opin. Ther. Targets 20222631933210.1080/14728222.2022.206651835503226 PMC 9239432 · doi ↗ · pubmed ↗