SurvDB: Systematic Identification of Potential Prognostic Biomarkers in 33 Cancer Types
Zejun Wu, Congcong Min, Wen Cao, Feiyang Xue, Xiaohong Wu, Yanbo Yang, Jianye Yang, Xiaohui Niu, Jing Gong

TL;DR
SurvDB is a new database that identifies over 4 million potential cancer prognostic biomarkers across 33 cancer types using multi-omics data from TCGA.
Contribution
The study introduces SurvDB, a comprehensive database integrating nine molecular data types for multi-omics cancer prognosis analysis.
Findings
Over 4.4 million molecular biomarkers were identified as significantly associated with cancer prognosis.
The database integrates nine molecular data types across 33 cancer types and over 10,000 samples.
SurvDB supports interactive data retrieval, visualization, and download for precision oncology research.
Abstract
The identification of cancer prognostic biomarkers is crucial for predicting disease progression, optimizing personalized therapies, and improving patient survival. Molecular biomarkers are increasingly being identified for cancer prognosis estimation. However, existing studies and databases often focus on single-type molecular biomarkers, deficient in comprehensive multi-omics data integration, which constrains the comprehensive exploration of biomarkers and underlying mechanisms. To fill this gap, we conducted a systematic prognostic analysis using over 10,000 samples across 33 cancer types from The Cancer Genome Atlas (TCGA). Our study integrated nine types of molecular biomarker-related data: single-nucleotide polymorphism (SNP), copy number variation (CNV), alternative splicing (AS), alternative polyadenylation (APA), coding gene expression, DNA methylation, lncRNA expression,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
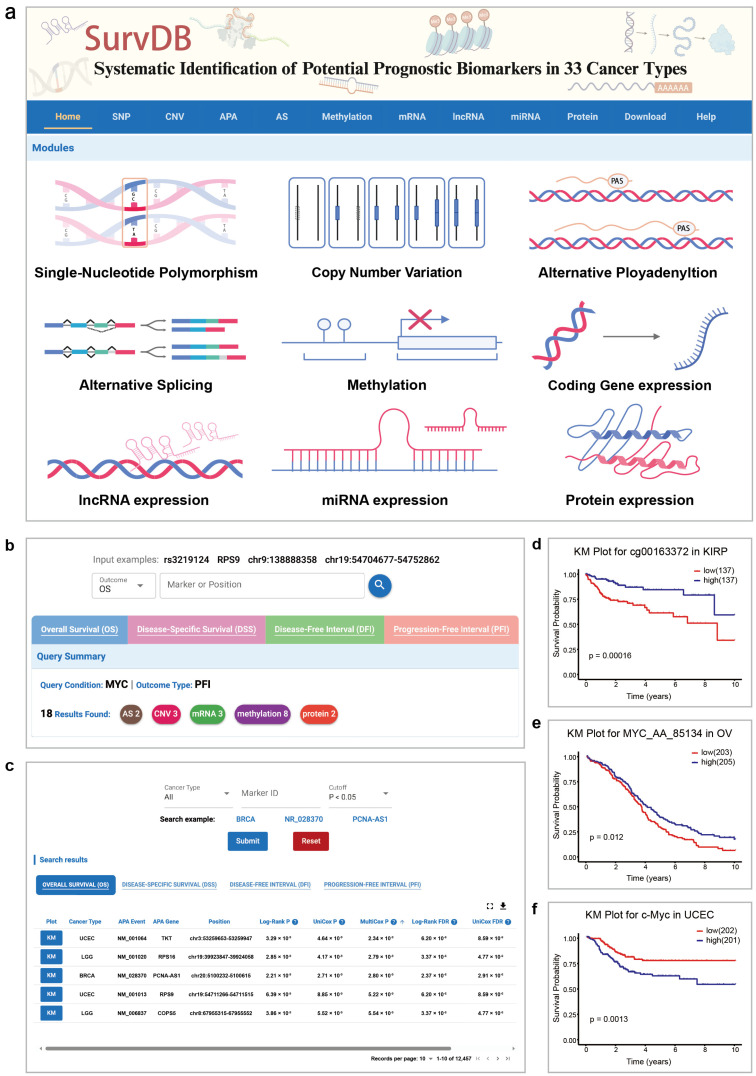 Figure 1
Figure 1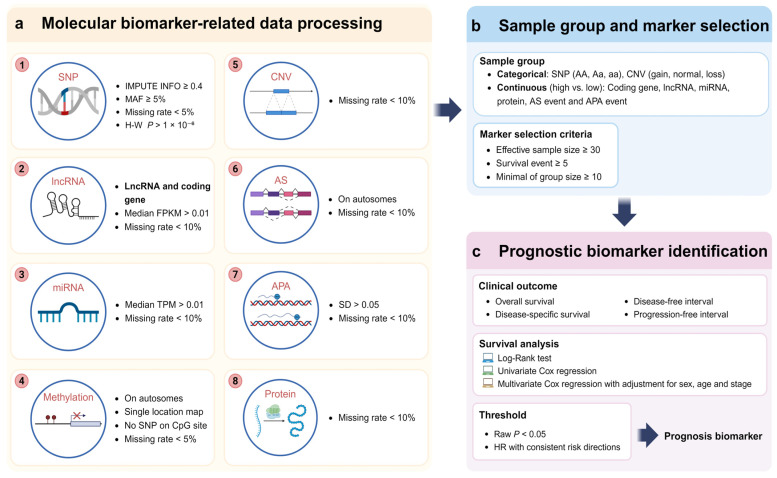 Figure 2
Figure 2- —Fundamental Research Funds for the Central Universities
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRNA modifications and cancer · Ferroptosis and cancer prognosis · Cancer-related molecular mechanisms research
1. Introduction
Cancer is a leading cause of global mortality [1]. According to the World Health Organization (WHO), approximately 20 million new cases and 9.7 million deaths reported worldwide in 2022 [2]. Precision therapy based on cancer prognostic biomarkers can significantly extend survival and improve the quality of life for cancer patients [3].
Cancer prognosis refers to the estimation of patient endpoints, including survival duration, recurrence risk, or disease progression after diagnosis [4]. Clinical outcome endpoints include overall survival (OS), disease-specific survival (DSS), disease-free interval (DFI), and progression-free interval (PFI) [5]. OS measures the time from diagnosis or treatment to death from any cause, providing a general survival assessment. DSS measures the time from diagnosis or treatment to death specifically caused by the disease, providing a clearer assessment of disease-specific lethality. DFI measures the time from complete remission (e.g., no residual tumor after surgery) to disease recurrence, assessing the duration of a disease-free state. PFI measures the time from stable disease to biological progression. While OS and DSS focus on survival duration, DFI and PFI emphasize the evaluation of disease status, as DFI highlights recurrence after a disease-free state and PFI tracks disease progression under controlled conditions, making them more critical for assessing the underlying dynamics of disease than OS and DSS [6,7]. In precision medicine, DFI and PFI have gained increasing attention as indicators of treatment efficacy, as they reflect the effects of therapy on disease control, enabling clinicians to promptly adjust treatment plans and achieve the goals of precision medicine. Moreover, given the relatively short clinical follow-up records for most of The Cancer Genome Atlas (TCGA) cohorts, PFI and DFI might generally be considered better endpoints choices than OS and DSS [8]. However, to our knowledge, current databases lack sufficient focus on PFI and DFI.
Previous studies have revealed significant variations in survival across cancer types. For instance, thyroid carcinoma (THCA) exhibits a relatively high five-year survival rate of 92.9%, in contrast to pancreatic adenocarcinoma (PAAD), which has a markedly lower rate of 8.5% [9]. Even within the same cancer type, OS varies considerably. In TCGA database, the median OS for skin cutaneous melanoma (SKCM) is 36.4 months, yet 14.7% of cases survive over 10 years. These results show the high heterogeneity of cancer prognosis. Investigating factors influencing cancer prognosis is essential for understanding cancer progression mechanisms and can inform clinical decision making and treatment efficacy evaluation [8,10]. For instance, in HER2-positive breast cancer, trastuzumab-based combination therapy significantly improves patient survival and quality of life [11,12].
With advancements in sequencing technologies and the increasing demands of precision medicine, cancer prognosis biomarker research has shifted from traditional clinical and demographic indicators to molecular level precision and personalized biomarker assessments [13,14]. Numerous omics data, including genomics, transcriptomics, and proteomics, are increasingly used for prognostic analysis [15,16,17]. Various biomarkers from different omics layers, such as single-nucleotide polymorphism (SNP), DNA methylation, and gene expression level, have been associated with cancer prognosis. For instance, the SNP rs27770A > G variant in the PLK1 has been reported to reduce its binding affinity to miRNA, suppressing mRNA expression and influencing liver hepatocellular carcinoma (LIHC) prognosis [18]. Biomarkers such as TP53 [19] and PD-L1 [20] have also been linked to multiple cancer prognoses and demonstrate promising potential in clinical applications. Copy number variations (CNVs) have been used to estimate some cancer progression [21,22]. Long non-coding RNA (lncRNA) is used to evaluate the diagnosis and treatment of non-triple-negative and triple-negative breast cancer and other cancer [23,24]. Alternative polyadenylation (APA) might lead to a worse prognosis in some cancers [25]. In cancer, aberrant alternative splicing (AS) patterns are frequently observed and known to contribute to carcinogenesis, de-differentiation, and metastasis [26]. Aberrant DNA methylation has been observed in various human diseases, including cancer [27]. However, to our knowledge, existing studies and databases are often limited to single-type molecular biomarkers for cancer prognosis.
TCGA database provides clinical and multi-omics data, including genomic, transcriptomic, proteomic, and methylation data, for over 10,000 samples across 33 cancer types, offering a valuable resource for cancer prognosis research. Several prognostic databases have been developed based on TCGA, such as GEPIA2 [28], TCPA [29], SurvivalMeth [30], OncoSplicing [31], and OSppc [32]. However, these databases primarily focus on single-type molecular biomarkers and lack comprehensive integration and analysis capabilities for multiple biomarker types, limiting the exploration of the complex molecular mechanisms underlying cancer prognosis. Additionally, some types of molecular data like genomic variant, CNV, APA, AS, and miRNA expression remain underexplored in prognostic studies. Furthermore, many databases assess only 1–2 clinical outcomes, such as OS or DSS, while neglecting DFI and PFI. To address these limitations, we systematically analyzed the relationship between nine types of molecular data and four clinical outcomes (OS, DSS, PFI, DFI). Additionally, we developed a comprehensive cancer prognosis database SurvDB (https://gong_lab.hzau.edu.cn/SurvDB/, accessed on 1 January 2025), an interactive online database for data retrieval, visualization, and download. SurvDB provides a comprehensive resource for candidate biomarker discovery and precision oncology research, and will support the exploration of complex molecular mechanisms underlying cancer prognosis.
2. Results
2.1. Data Summary of SurvDB
In SurvDB, we used multi-omics and clinical data from 33 cancer types available in TCGA database, encompassing 11,160 tumor samples. The sample size for each cancer type ranged from 12 in uveal melanoma (UVM) to 1207 in breast invasive carcinoma (BRCA) (Table 1).
From TCGA and its derivative databases, we integrated nine types of molecular biomarker-related data: SNP, CNV, AS, APA, coding gene expression, DNA methylation, lncRNA expression, miRNA expression, and protein expression. To ensure consistency and simplify subsequent descriptions, all molecules or indices within these datasets are collectively referred to as “markers”. In total, we analyzed the relationship between 6,867,129 markers and cancer prognosis (Table 2). Of them, the SNP, CNV, mRNA expression, lncRNA expression, miRNA expression and DNA methylation data were directly downloaded from TCGA database, while the Percentage of Distal polyA site Usage Index (PDUI) data for APA events, percent spliced index (PSI) data for AS events, and protein expression data were downloaded from TC3A database, TCGA SpliceSeq, and TCPA database, respectively [29,33,34].
After filtering by imputation score, minor allele frequency (MAF), missing rate, and Hardy–Weinberg p-value, an average of 3,352,031 SNPs per cancer type were retained. For mRNA, lncRNA, and miRNA, after removing low-expression genes (FPKM < 0.01 for mRNA and lncRNA, TPM < 0.01 for miRNA), an average of 16,894 coding genes, 7634 lncRNAs, and 480 miRNAs were retained per cancer type. After quality control, an average of 3846 APA events, 25,937 alternative splicing (AS) events, 366,644 DNA methylation sites, 19,629 CNVs and 219 proteins per cancer type were retained for downstream analysis.
Potential prognostic biomarkers for nine types of molecular data across 33 cancers were identified through Log-rank test, uni-Cox, and multi-Cox, with p < 0.05 as the significance threshold. A total of 4,498,523 unique prognostic biomarkers were identified across four clinical outcomes, encompassing nine types of molecular data. (Table 3), including 4,035,082 SNPs, 17,730 coding genes, 9837 lncRNAs, 851 miRNAs, 446 proteins, 367,478 methylation sites, 35,695 AS events, 6942 APA events, and 24,462 CNVs.
2.2. Database Construction
All results were stored in a MongoDB database (v3.4.2). A user-friendly web interface, SurvDB (https://gong_lab.hzau.edu.cn/SurvDB/, accessed on 1 January 2025), was developed using the Flask framework (v1.0.3) to support data browsing, searching, and downloading. The database operates on an Apache2 web server (v2.4.18) and is compatible with multiple browsers across various operating systems.
2.3. Functions and Usage of SurvDB
SurvDB provides a user-friendly web interface for users to browse, visualize, search, and download prognostic biomarkers of different types (Figure 1). To fully utilize multi-omics data, we designed an aggregation query module. By entering a marker name or a genomic region, users can obtain integrated search results, including all related information across multiple cancers or types of molecular markers. For example, multiple genetic loci, APA, and CNV in the chromosomal region 9q21.3 (chr9:21800000-22400000) have been reported associated with various cancers [35]. Users can input chr9:21800000-22400000 to retrieve all potential prognostic biomarkers identified across different types of molecular data in that region. Additionally, multiple types of molecular markers are linked with genes, and the results for these genes across four clinical outcomes are shown. By entering a specific gene, users can obtain related molecular markers associated with four clinical outcomes. For example, for the MYC gene, 15 results are shown in OS section. As for other three outcomes (DSS, DFI, PFI), there are 17, 11, and 18 results. In PFI section, the 18 results include 8 methylation records, 3 records each for mRNA and CNV, and 2 records each for AS and protein (Figure 1b).
On the separate query page for each type of molecular data (Figure 1c), users can query analysis results for individual markers based on cancer type, marker ID, and genomic location. The Kaplan–Meier (KM) plotter shows examples of methylation, AS, and protein results in MYC search results, respectively (Figure 1d–f). The “Help” page offers database descriptions and usage guides, and feedback can be emailed using the address at the page’s bottom.
3. Discussion
This study utilized multi-omics data provided by TCGA database and systematically analyzed nine types of molecular data to identify potential prognostic biomarkers by combining three classic survival analysis methods: Log-rank test, uni-Cox, and multi-Cox. Additionally, to better present the results, we developed a user-friendly SurvDB database to facilitate querying, browsing, and downloading by users.
Compared to other cancer prognosis-related databases, SurvDB offers the following advantages. First, survdb incorporates more types of molecular markers, such as genomic variations, CNV, APA, and miRNA expression. Research has shown that APA of the CSTF2 is associated with lung cancer prognosis [36], and miRNA-21 is related to the prognosis of metastatic colorectal cancer [37]. A systematic analysis of these types of molecular markers and their relationship with cancer prognosis will provide more insights for further biological experiments and clinical research.
The integration of multi-omics data offers a new perspective for cancer prognosis research. By combining various omics data, such as gene expression, mutation, and epigenetic information, it is possible to gain a more comprehensive understanding of the complexity of cancer and improve the accuracy of prognosis prediction. For instance, a study based on TCGA constructed a lung adenocarcinoma prognosis-related risk prediction model by integrating multi-omics data, demonstrating the potential of multi-omics data in improving prognosis accuracy [38]. Additionally, a study on liver cancer, published by the collaborative team of Tsinghua University, also highlighted the value of multi-omics data in cancer prognosis research [39]. The prognostic findings of multiple types of molecular markers in this study offer insights for multi-omics feature selection.
This study also identified different types of molecular biomarkers associated with DFI and PFI, addressing the lack of focus on clinical outcome endpoints in other databases. These findings contribute to the mechanistic exploration of DFI and PFI. SurvDB also features a multi-cancer and multi-molecule joint query function, which helps other researchers conduct multi-level mechanistic investigations.
The integrative identification of prognostic biomarkers enhances understanding of cancer progression and aids in identifying high-risk patients, offering valuable guidance for clinical decision making. Cancer recurrence is a specific prognostic outcome. For example, Professor Luo’s team from Sun Yat-sen University [40] combined TCGA renal cancer data with data from 227 Chinese patients and used the multicenter retrospective analysis method to identify six SNPs closely associated with localized renal cancer recurrence in Chinese populations. They further demonstrated that integrating these six SNPs into a predictive model alongside clinical pathological indicators improved prediction accuracy, enabling the more precise identification of high-risk patients for recurrence. They proposed that intensified monitoring and adjuvant therapy for high-risk patients could mitigate adverse outcomes.
In the future, we will explore artificial intelligence and machine learning techniques to fully utilize the identified biomarkers for constructing multi-omics predictive models, aiming to improve the accuracy of prognosis prediction and its clinical applicability. Additionally, this research has revealed significant prognostic variability across populations, while TCGA data mainly represent Western cohorts [41]. Thus, we aim to expand the SurvDB database by incorporating data from diverse populations, additional cancer subtypes, and broader biomarker categories. Ultimately, we hope that SurvDB will become a vital resource for cancer prognosis researchers, promoting advancements in cancer prognosis studies and precision medicine.
4. Materials and Methods
4.1. Molecular Data Collection and Processing
Nine types of molecular biomarker-related data were collected from TCGA and its derivative databases, followed by processing and quality control (Figure 2). Genotype data detected using the Affymetrix SNP 6.0 array were obtained from TCGA. We imputed autosomal variants in all samples for each cancer type using IMPUTE2 (v2.3.2), with the 1000 Genomes Phase 3 as the reference panel [42]. The imputation was performed in the two-step procedure provided by IMPUTE2. After imputation, SNPs were filtered using the following criteria: (i) imputation quality score ≥ 0.4, (ii) minor allele frequency (MAF) ≥ 5%, (iii) missing rate < 5%, and (iv) Hardy–Weinberg equilibrium p-value > 1 × 10^−6^.
For the quality control of coding gene and lncRNA, we downloaded the gene expression profile from TCGA and excluded all the genes with an extremely low expression median fragment per kilobase million (FPKM) < 0.01 and a missing rate > 10% for downstream analysis. Then, genes were classified into coding gene and lncRNA according to the annotation from ENCODE (v36) [26,27].
For the quality control of miRNA, we downloaded the miRNA sequencing data from TCGA and excluded the miRNAs with a median transcription per million (TPM) < 0.01 and those with missing rate > 10%.
PSI, a commonly used metric for quantifying splicing events, is defined as the ratio of reads indicating the presence of a transcript element versus the total number of reads covering the splicing event. After downloading PSI data from TCGA SpliceSeq [34], we excluded AS events with a missing rate > 10% across all samples or those located on sex chromosomes.
PDUI quantifies the alternative polyadenylation (APA) frequency based on the relative usage of distal polyA sites. The PDUI data were obtained from TC3A database [33]. APA events with a missing rate >10% or a standard deviation < 5% were excluded.
DNA methylation data were obtained from TCGA database, generated using the Illumina Infinium HumanMethylation450 BeadChip array. We downloaded these data from TCGA data portal and filtered out the sites according to the following criteria: (i) on sex chromosomes; (ii) mapping to multiple locations on the genome; (iii) containing known SNP on CpG sites; and (iv) beta value with a missing rate > 5%.
CNV data were retrieved from TCGA and further processed using GISTIC2.0 to discretize CNV states into five categories: homozygous deletion (−2), single-copy loss (−1), diploid (0), low-level gain (1), and high-level amplification (2) [43]. CNV data were mapped to human genome coordinates using the HUGO probeMap from UCSC Xena to determine the copy number state for each gene [44].
Protein expression data were derived from reverse-phase protein array (RPPA) experiments available in TCGA. Expression levels for 282 proteins were obtained from TCPA database and further annotated at gene level.
4.2. Clinical Data Collection and Processing
Clinical data were obtained from TCGA database, including patient age, sex, tumor stage, OS, DSS, DFI, and PFI. Redundant samples from the same patient were excluded. For each cancer type, only patient samples with complete clinical information were included in the analysis.
4.3. Identification of Prognostic Biomarkers
During the identification of genotype- and CNV-related prognostic biomarkers, samples were grouped based on their classification. To ensure the reliability of subsequent survival analysis results, markers with fewer than 30 valid samples or fewer than 5 samples in the smallest group were excluded from the analysis. For other types of molecular markers, in each analysis, samples were grouped into two groups based on the median value of the marker. Next, we employed three classic survival analysis methods, Log-rank test, uni-Cox, and multi-Cox, to systematically assess the correlation between markers and cancer patient outcomes, including OS, DSS, PFI, and DFI. The multi-Cox model further incorporated covariates such as gender, diagnosis age, and tumor stage to adjust for potential confounders.
As a database, we aim to retain more information on significant finding. Therefore, markers that were consistently significant (raw p-value < 0.05) and exhibited consistent risk directions across all methods were selected as possible prognostic biomarkers.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Bray F. Laversanne M. Weiderpass E. Soerjomataram I. The ever-increasing importance of cancer as a leading cause of premature death worldwide Cancer 20211273029303010.1002/cncr.3358734086348 · doi ↗ · pubmed ↗
- 2Bray F. Laversanne M. Sung H. Ferlay J. Siegel R.L. Soerjomataram I. Jemal A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries CA Cancer J. Clin.20247422926310.3322/caac.2183438572751 · doi ↗ · pubmed ↗
- 3Rizk E.M. Gartrell R.D. Barker L.W. Esancy C.L. Finkel G.G. Bordbar D.D. Saenger Y.M. Prognostic and predictive immunohistochemistry-based biomarkers in cancer and immunotherapy Hematol. Oncol. Clin. N. Am.20193329129910.1016/j.hoc.2018.12.005PMC 649706930833001 · doi ↗ · pubmed ↗
- 4Busund M. Ursin G. Lund E. Chen S.L.F. Rylander C. Menopausal hormone therapy and incidence, mortality, and survival of breast cancer subtypes: A prospective cohort study Breast Cancer Res.20242615110.1186/s 13058-024-01897-439497219 PMC 11536865 · doi ↗ · pubmed ↗
- 5Jiang J. Chen Z. Wang H. Wang Y. Zheng J. Guo Y. Jiang Y. Mo Z. Screening and identification of a prognostic model of ovarian cancer by combination of transcriptomic and proteomic data Biomolecules 20231368510.3390/biom 1304068537189432 PMC 10136255 · doi ↗ · pubmed ↗
- 6Sakamaki Y. Ishida D. Tanaka R. Prognosis of patients with recurrence after pulmonary metastasectomy for colorectal cancer Gen. Thorac. Cardiovasc. Surg.2020681172117810.1007/s 11748-020-01368-532323124 · doi ↗ · pubmed ↗
- 7Morra S. Scheipner L. Baudo A. Jannello L.M.I. de Angelis M. Siech C. Goyal J.A. Touma N. Tian Z. Saad F. Contemporary conditional cancer-specific survival rates in surgically treated nonmetastatic primary urethral carcinoma J. Surg. Oncol.20241291348135310.1002/jso.2763738606531 · doi ↗ · pubmed ↗
- 8Liu J. Lichtenberg T. Hoadley K.A. Poisson L.M. Lazar A.J. Cherniack A.D. Kovatich A.J. Benz C.C. Levine D.A. Lee A.V. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics Cell 201817340041610.1016/j.cell.2018.02.05229625055 PMC 6066282 · doi ↗ · pubmed ↗