PAR2 Serves an Indispensable Role in Controlling PAR4 Oncogenicity: The β-Catenin–p53 Axis
Priyanga Appasamy, Jeetendra Kumar Nag, Hodaya Malka, Rachel Bar-Shavit

TL;DR
This study shows that PAR2 is essential for PAR4's cancer-promoting effects, and a compound called Pc(4-4) can inhibit this process by boosting p53 activity.
Contribution
The study reveals a novel role of PAR2 in modulating PAR4 oncogenicity through the β-catenin–p53 axis and identifies Pc(4-4) as a potential anti-cancer drug.
Findings
PAR2 knockdown or TrPAR2 expression inhibits PAR4-induced β-catenin stabilization and tumor growth.
Pc(4-4) inhibits PAR2 activity by increasing p53 and p21 levels, reducing cancer cell invasion and migration.
PAR2 signaling is indispensable for PAR4's pro-tumor functions.
Abstract
Although the role of G-protein-coupled receptors (GPCRs) in cancer is acknowledged, GPCR-based cancer therapy is rare. Mammalian protease-activated receptors (PARs), a sub-group of GPCRs, comprise four family members, termed PAR1–4. Here, we demonstrate that PAR2 is dominant over PAR4 oncogene in cancer. We performed a knockdown of Par2/f2rl1 and expressed C-terminally truncated PAR2 (TrPAR2), incapable of inducing signaling, to assess the impact of PAR2 on PAR4 oncogenic function by β-catenin stabilization assessment, immunoprecipitation, and xenograft tumor generation in Nude/Nude mice. PAR2 and PAR4 act together to promote tumor generation. Knockdown Par2 and TrPAR2 inhibited the PAR2 and PAR4-induced β-catenin levels, nuclear dishevelled 1(DVL1), and TOPflash reporter activity. Likewise, PAR2 and PAR4-induced invasion and migration were inhibited when Par2 was knocked down or in the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
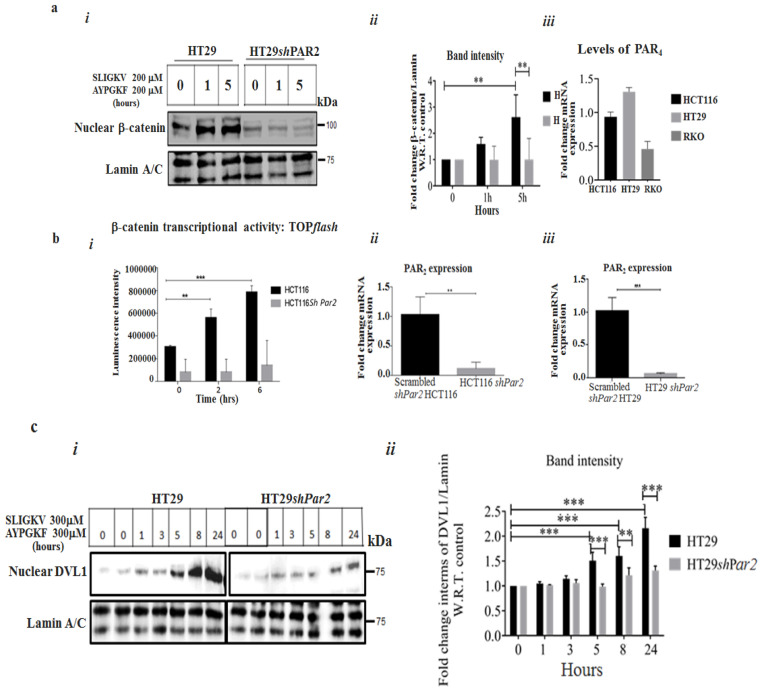 Figure 1
Figure 1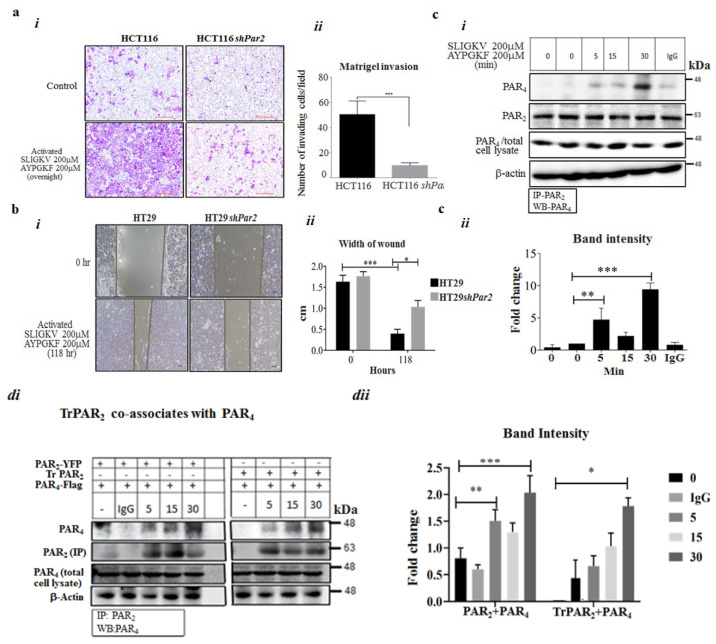 Figure 2
Figure 2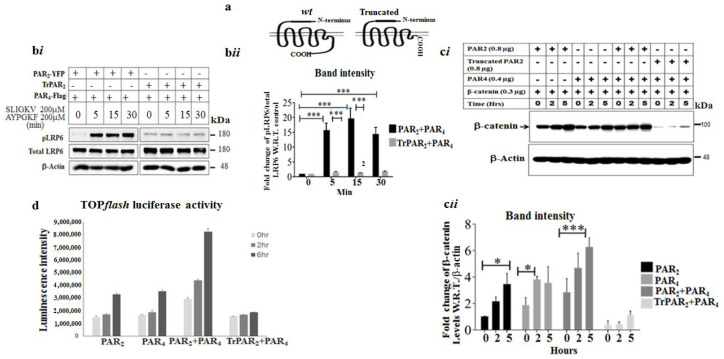 Figure 3
Figure 3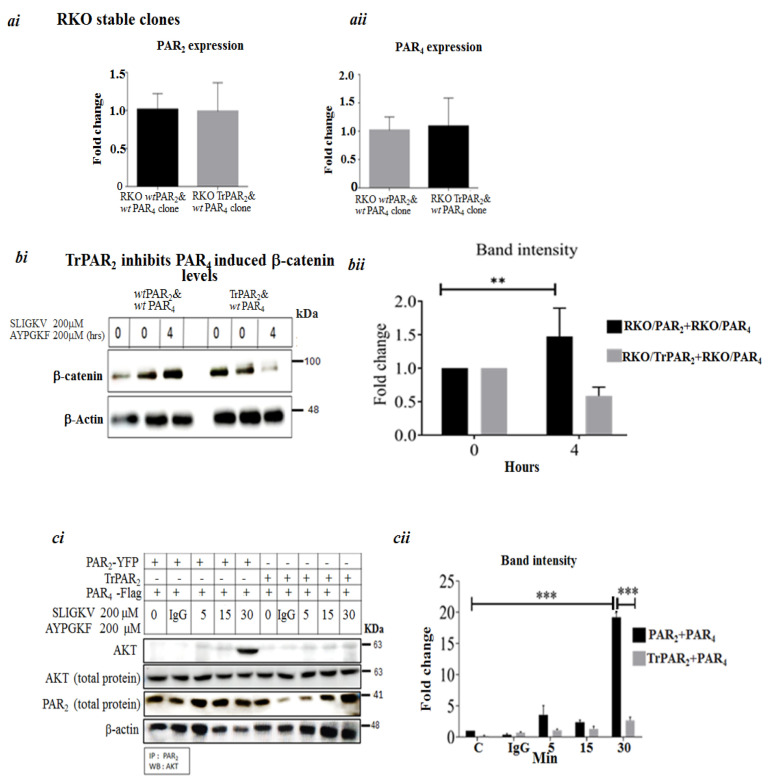 Figure 4
Figure 4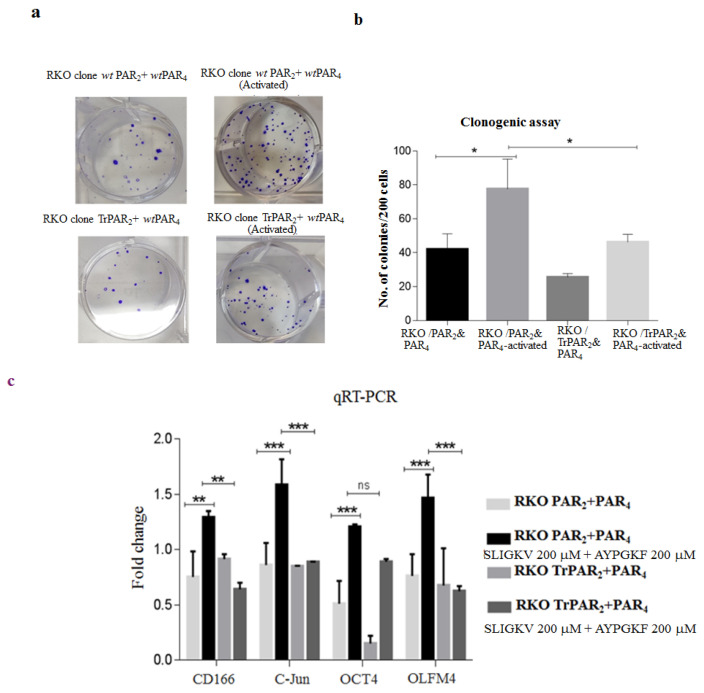 Figure 5
Figure 5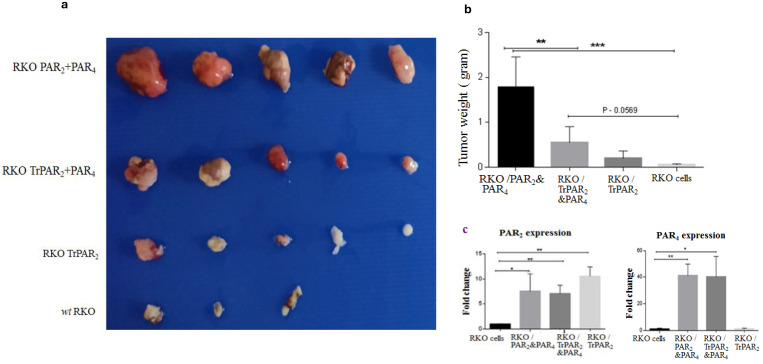 Figure 6
Figure 6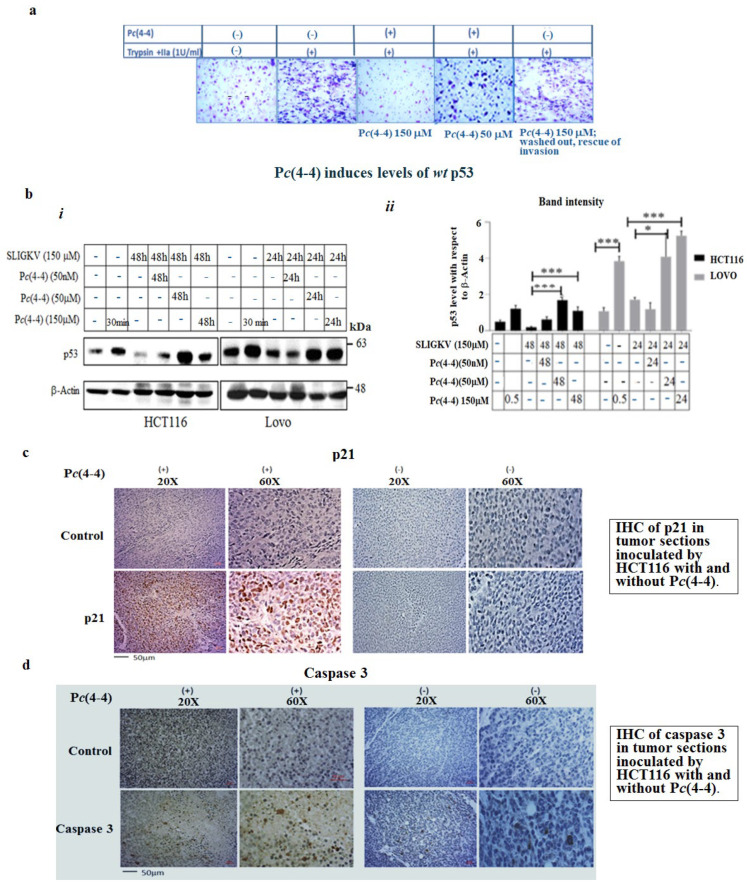 Figure 7
Figure 7- —Israel Science Foundation
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCancer-related gene regulation · Cell Adhesion Molecules Research · RNA Research and Splicing
1. Introduction
Despite an increasing understanding of G-protein-coupled receptor (GPCR)-facilitated cancer pathogenesis, little is known about the role of GPCRs in epithelial malignancies. GPCRs comprise a diverse super-family of proteins, which serve as biological targets for pharmaceutical drug design. GPCR-targeted drugs presently represent nearly 30% of all therapeutics directed against a wide range of pathologies. Yet, nearly no GPCR-based drugs are clinically used in cancer [1,2,3].
In the United States (US) alone during 2024, over 2 million (e.g., 2,001,140) new cancer cases were diagnosed and over 600,000 (e.g., 611,720) cancer deaths were reported [4,5]. In fact, the 5-year survival rate for cancer in general has increased from 49% through the mid 1970s to 69% in 2013–2019. This achievement is mainly due to earlier detection, reduced smoking, and the advanced treatments, resulting in over 4 million deaths prevented since 1991.
Frizzled (FZD) receptors, a sub-group of GPCRs, are activated by Wnt ligands to initiate the canonical Wnt/β-catenin pathway. The Wnt/β-catenin signaling pathway regulates embryonic development, tissue homeostasis, and cancer [6]. Once bound to a complex comprising FZD and low-density lipoprotein receptor protein (LRP)5/6 coreceptors, Wnts initiate the β-catenin signaling pathway. This leads to the detachment of β-catenin from its cellular degradation complex, promoting β-catenin stabilization and nuclear translocation, where β-catenin acts as a transcription factor, inducing the level of a specific gene signature. As part of this process, Disheveled (DVL), a cytoplasmic adaptor protein that connects FZD to downstream components, enters the nucleus and becomes part of the transcription complex. A negative layer of regulation is provided by both RNF43 and ZNRF3, ubiquitin ligases that stimulate the degradation of FZD and LRP5/6 co-receptors. As a result of such degradation, membrane receptor availability and downstream β-catenin signaling are markedly reduced [7,8].
Mammalian protease-activated receptors (PARs) correspond to another sub-group of GPCRs and comprise four family members (PAR_1–4_), all of which are uniquely activated via proteases [9]. Proteases residing within the tumor microenvironment are either immobilized to the extracellular basement membranes as a depot storage site or, when found in a soluble form, are involved in the activation of PARs, contributing to the maintenance of tumor growth and progression. PAR_1,3,4_ are thrombin receptors and PAR_2_ is a trypsin receptor. Noticeably, PAR_2_ and PAR_4_ can be activated by the same protease. MAPkinase signaling is also involved in PAR-induced tumor growth and progression [9]. We have previously demonstrated that PAR members potently induce β-catenin stabilization, leading to β-catenin nuclear translocation and transcriptional activity [10,11,12,13]. In accordance, we have recently shown that the E3 ubiquitin ligase RNF43 negatively regulates PAR_2_ cell-surface levels and consequently, PAR_2_-induced β-catenin signaling, similar to the actions of RNF43 on FZDs. Likewise, PAR_2_ degradation is rescued by RSPO-LGR5 axis [14]. Hence, it is proposed that PARs GPCRs play a role in the β-catenin stabilization cell signaling.
Advanced bioinformatics tools and DNA sequencing have enabled the characterization of the tumor gene landscape. This has allowed for high-throughput RNA sequence analyses of selected GPCR transcriptional profiles, revealing the expression of 195 GPCRs upon cell reprogramming, leading to cancer-stem-cell (CSC) sphere formation. It was shown that PAR_4_ (f2rl3) and PAR_2_ (f2rl1) are considerably up-regulated upon CSC sphere formation [15]. In other studies, PAR_4_ has emerged as a potent oncogene that is over-expressed in cancer epithelial cells and capable of inducing tumors in vivo [16,17,18,19,20]. We have previously demonstrated that PAR_4_ is a potent oncogene capable of inducing tumors in a xenograft mouse model, in vivo [21]. In addition, we have shown earlier that Pc(4-4), a lead backbone cyclic peptide [22,23,24] selected out of a cyclic peptide library directed toward PAR_2&4_ pleckstrin homology (PH)- binding motifs [21,25,26], effectively inhibited tumor growth.
Hierarchy exists within the PAR family. We previously demonstrated that PAR_1_-promoted cancer processes require the presence of PAR_2_. This was shown using either a shPar2 knockdown of PAR_2_ expression or by the use of a truncated form of PAR_2_ lacking the entire cytoplasmic tail, a variant incapable of promoting cell signaling [27]. This concurs with studies by Sevigny et al. [28], demonstrating that PAR_2_ affects PAR_1_ function in neointimal arterial thickening of smooth muscle cell (SMC) growth. It also supports previous work from this group and others on PAR_1_ and PAR_2_ trans-activation [29,30]. We now ask what the inter-relations between PAR_2_ and PAR_4_ in cancer are.
Indications from various tumor models propose that the Wnt signaling and p53 pathways collaborate to promote tumor growth and progression [31,32]. Indeed, there is a cross-talk between β-catenin and p53 in cancer. Data based on genetic analysis of colorectal cancer (CRC) patients showed induced β-catenin stabilization as a result of mutated APC and β-catenin, together with a high incidence of p53 mutations [33]. In mice carrying mutant p53 (p53^R270H^), increased intestinal tumor growth and a rise in invasiveness were observed [34]. Moreover, the transformation of normal colonic stem cells via the accumulation of mutated APC, KRAS, and SMAD4 genes failed to metastasize in the presence of wt p53, as occurs in CRC [35,36]. At the same time, the full malignant repertoire of invasive CRC is obtained in the presence of mutated p53^R270H^ [34]. In lung cancer, for example, p53 mutations impact personalized therapy toward epidermal growth factor receptor (EGFR) mutants [37,38].
Indeed, only upon the wt p53 pathway does de-regulated β-catenin fully manifest its oncogenic properties. This also occurs reciprocally, whereby increased levels of wt p53 inhibit de-regulated β-catenin in a variety of cell settings. Therefore, a negative feedback loop links wt p53 and β-catenin, with an interruption of this loop most likely affecting oncogenic β-catenin tumorigenesis [39].
In the present study, our overall aim was to examine functional interactions of PAR_2_ and PAR_4_ in colon cancer. This was assessed via PAR-induced β-catenin-signaling events, PAR-PH-Akt association, colony formation, Matrigel invasion in vitro, and tumor development in vivo. In the presence of either knockdown Par2 or truncated (Tr) PAR_2_ lacking the cytoplasmic tail, a marked inhibition of PAR_2&4_ signaling events was observed. This indicates the essential role of PAR_2_ signaling in PAR_4_-induced tumor growth. Furthermore, a Pc(4-4) compound directed toward the PAR PH-binding motif [21,25] was shown to act through an increase in the level of wt p53. This highlights Pc(4-4) as a promising anti-cancer drug.
2. Results
2.1. Knockdown of Par2/f2rl1 Inhibits Events in PAR2 and PAR4-Induced β-Catenin Stabilization and Cell Invasion
Upon the knockdown of Par2 alone, the inhibition of the induced β-catenin level signaling was obtained (Figure 1a(i),a(ii)). High expression levels of both Par2/f2rl1 and Par4/f2rl3 (in addition to other oncogenes) were observed in HCT116 and HT29 cells (Figure 1a(iii)). Significantly, while we previously demonstrated evidence of the potent individual activation of β-catenin stabilization [12,14,40] by PAR_2_ via the addition of SLIGKV peptide or by PAR_4_ via AYPGKF peptide, we now aimed to elucidate the relative impact of PAR_2_ on PAR_2_ and PAR_4_ function. For the assessment of the relative impact of Par2/f2rl1 on pro-tumor signaling, we knocked down Par2 using shRNA-Par2 in colon-cancer-cell lines (i.e., HCT116 and HT29). Decreased Par2 levels were observed, compared with those in scrambled shPar2-infected cells (Figure 1b(ii),b(iii)). When combined, the SLIGKV and AYPGKF peptides induced the activation of PAR_2_ [12] and PAR_4_, leading to induced β-catenin stabilization. Along this line, an increased TOPflash reporter activity of β-catenin transcription function was obtained following PAR_2_ and PAR_4_ activation. When Par2 was knocked down, a marked inhibition of PAR_2_ and PAR_4_-induced TOPflash activity was observed (Figure 1b). When levels of β-catenin were evaluated in HT29 cells, high β-catenin levels were seen following peptide SLIGKV- and AYPGKF-mediated PAR_2_ and PAR_4_ activation. Upon the silencing of Par2 RNA levels (it is assumed that under these conditions, levels of their respective protein levels are reduced accordingly, although not measured directly) in HT29 cells, a marked inhibition of β-catenin level was seen (Figure 1a(i)–(iii)). The activation of PAR_2_ induced DVL localization into the cell nucleus. Nuclear DVL now becomes part of a transcriptional complex composed, among others, of c-Jun, β-catenin, and Tcf [41]. While abundant nuclear DVL1 levels were observed following the activation of both PAR_2_ and PAR_4_, (Figure 1c(i),c(ii)), upon Par2 knockdown, a significant inhibition of nuclear DVL1 level was obtained. Overall, these results point to the essential requirement of PAR_2_ expression for PAR_2_ and PAR_4_-induced β-catenin stabilization events.
The activation of PAR_4_ and PAR_2_ enhanced the invasion of HCT116 cells through Matrigel-coated filter membranes. This stands in contrast to the marked inhibition of PAR_2_ and PAR_4_-induced invasion seen when Par2 was knocked down (Figure 2a). Similarly, PAR_2_ and PAR_4_ activation resulted in a covering of the space generated in a wound-scratch assay (Figure 2b). When Par2 was silenced, cell proliferation/migration in the assay was potently inhibited.
2.2. Co-Association of PAR2 and PAR4
It was suggested that PAR_2_ and PAR_4_ are localized in proximity, thereby enabling their action as a single functional unit upon activation. To obtain direct evidence for PAR_2_–PAR_4_ heterodimer formation, we performed co-immunoprecipitation (co-IP) analysis. For this purpose, wt Par2 and wt Par4 were ectopically over-expressed in HEK293 cells. The cells were treated with both the SLIGKV and AYPGKF peptides for various intervals and were further processed to obtain cell lysates. Next, IP was carried out using anti-PAR_2_ antibodies, with IgG serving as a control. As shown, co-association between PAR_2_ and PAR_4_ was observed after 5 and 10 min, reaching maximal association after 30 min of activation of both receptors (Figure 2c). This result supports our notion that PAR_2_ acts in conjunction with PAR_4_, forming a PAR_2_–PAR_4_ complex that can be observed as soon as 5 min after activation and for up to 30 min.
Similarly, a truncated form of PAR_2_, TrPAR_2_, lacking the cytoplasmic tail, is capable of co-associating with PAR_4_, as shown by the co-IP-based capture of PAR_4_ and TrPAR_2_ (Figure 2d(i),d(ii)). Co-association was obtained after the transient transfection of HEK293 cells with plasmids containing Par4/f2rl3 and TrPar2/f2rl1, as compared with transfection with plasmid containing both wt Par4/f2rl3 and wt Par2/f2rl1. Maximal co-association was seen after 30 min of activation of both PAR_2&4_ following activation induced by the AYPGKF and SLIGKV peptides.
2.3. TrPAR2 Inhibits PAR2&4-Induced β-Catenin Stabilization, Transcriptional Activity, Colony Formation and Stem-Cell Marker Levels
We next considered the effect of TrPAR_2_ on PAR_2_ and PAR_4_-induced tumor events in vitro and in vivo. The presence of a TrPAR_2_ lacking the cytoplasmic tail (Figure 3a), and hence incapable of promoting signaling, stressed the significance of PAR_2_-induced signaling events in PAR_4_ and PAR_2_-mediated activities. We transfected HEK293 cells with the TrPar2/f2rl1 and wt Par4/f2rl3 plasmids, as well as flg-β-catenin constructs, and compared their functions with that of cells transfected with plasmids for wt Par2/f2rl1, Par4/f2rl3, and flg-β-catenin, whereas TrPAR_2_ was well expressed on the cell-surface membrane, as we previously reported [27], and a marked inhibition of LRP6 phosphorylation was seen. In contrast, robust LRP6 phosphorylation was observed following the activation of both PAR_2_ and PAR_4_ (Figure 3b(i),b(ii)). In addition to inhibiting pLRP6 levels, TrPAR_2_ also inhibited β-catenin levels. At the same time, a powerful enhancement of β-catenin levels was obtained following the transfection and activation of Par2/f2rl1 and/or Par4/f2rl3 either individually, or both. These enhanced β-catenin levels are potently inhibited in the presence of TrPAR_2_ (Figure 3c(i),c(ii)). Similarly, PAR-induced TOPflash β-catenin reporter activity observed following the activation of PAR_2_ and/or PAR_4_ is indicative of PAR-induced β-catenin transcriptional activity. In the presence of TrPAR_2_, the powerful inhibition of both PAR_2_- and PAR_4_-induced TOPflash activity was obtained (Figure 3d).
Next, we generated stable clones of RKO cells, a poorly differentiated colon-cancer-cell line, transformed on the background of mismatch repair. The clones generated over-expressed wt Par2/f2rl1 and wt Par4/f2rl3, separately and in combination of both PAR_2_ and PAR_4_, as well as TrPar2/f2rl1 and wt Par4/f2rl3 (Figure 4a). PAR-induced levels of β-catenin stabilization were then assessed in these clones.
While increased β-catenin levels were seen following SLIGKV and AYPGKF peptide-mediated activation of wt over-expressing clones, β-catenin levels were attenuated in clones over-expressing TrPAR_2_ and wt PAR_4_ (Figure 4b).
Previously, we identified PH-binding motifs within the PAR_2_ and PAR_4_ C-terminal tails that associate with PH-containing signal proteins, thus providing a powerful platform for drug design [21,25]. We now demonstrate that in the presence of TrPar2/f2rl1, the association between PAR and PH-Akt seen in both wt PAR_2_ and wt PAR_4_ was potently inhibited (Figure 4c). Such inhibition takes place despite TrPar2 being well expressed, as shown previously [27]. Together, these data support the conclusion that PAR_2_ signaling is required for PAR_4_ function.
A colony formation assay demonstrated the generation of abundant colonies upon activation of both PAR receptors. A marked inhibition of colony-forming ability was observed in the presence of the TrPAR_2_ variant and wt PAR_4_ (Figure 5a,b). Furthermore, elevated levels of stem-cell markers were seen upon activation of both PAR_2_ and PAR_4_ (Figure 5c). Reduced levels of these markers were obtained in the presence of TrPAR_2_ and wt PAR_4_ (Figure 5c). It is assumed that the levels of their proteins were inhibited accordingly, although not measured directly. Hence, the oncogenic activity of PAR_4_ is markedly inhibited when PAR_2_ is incapable of initiating cell signaling.
2.4. TrPAR2 Inhibits PAR2 and PAR4-Induced Tumor Growth In Vivo
To elucidate the dominant role of PAR_2_ over PAR_4_ in xenograft tumor growth in vivo, the following approach was taken. Stable RKO clones were inoculated subcutaneously into nude mice. The mice were monitored every other day for 35 days (or until the tumors became clearly noticeable), sacrificed, and the tumors were excised and embedded in paraffin. Whereas large tumors (e.g., ~1.2 cm) were observed in mice inoculated with the RKO clones over-expressing both wt PAR_2_ and wt PAR_4_, only a few to almost no tumors were seen following the injection of clones expressing TrPAR_2_ and wt PAR_4_. Likewise, only very small tumors were obtained when clones expressing either TrPar2 alone or wt RKO cells were inoculated into mice (Figure 6). The inhibition obtained was 4.5-fold higher for TrPAR_2_ and wt PAR_4_-generated tumors, as compared with wt PAR_2&4_-generated tumors. Together, both the in vitro and in vivo data obtained in the presence of a TrPAR_2_ indicated that TrPAR_2_ has a negative inhibitory effect on PAR_4_ and PAR_2_-induced tumor-promoting processes, similar to the inhibition seen following shRNA-mediated TrPar2/f2rl1 gene silencing.
2.5. The Oncogenic Properties of PAR2 Are Significantly Inhibited in the Presence of Pc(4-4) via Induced wt p53 Levels
We previously selected a Pc(4-4) compound from a library of cyclic peptides directed against the PAR_2_/or PAR_4_ PH-binding motif [21]. As we have shown, Pc(4-4) potently inhibited PAR-induced Akt association, cell migration, and invasion in vitro, as well as tumor generation in vivo [21,25]. PAR_2_ activity is necessary for PAR_4_ function (and also for PAR_1_; 27).
Now, we continue our assessment of the molecular mechanism of PAR_2_ by using Pc(4-4) as a potent inhibitor.
In order to assess the impact of Pc(4-4) on PAR induced tumorigenity that mimics physiological conditions by proteases activation, we performed a Matrigel invasion assay. It is shown that Pc(4-4) potently inhibits the Matrigel invasion of trypsin and thrombin (e.g., IIa)-activated PAR_2_ and PAR_4_, in HCT116 colon cancer cells (expressing both PAR_2_ and PAR_4_). The inhibition is observed between 50 nM and 150 μM of Pc(4-4). This inhibition is reversible, since upon extensive wash-out, increased Matrigel invasion is seen (Figure 7a).
We found that in the presence of Pc(4-4), a significant increase in the level of wt p53 was observed. As shown in Figure 7b, the level of wt p53 in aggressive colon-cancer-cell lines (e.g., HCT116 and Lovo) were rapidly down-regulated following PAR_2_ activation. In contrast, in the presence of Pc(4-4), sustained high levels of wt p53 were seen. This was shown for the lowest concentration of Pc(4-4) tested (50 nM), with wt p53 levels remaining elevated for 24 and 48 h, compared to untreated cells.
Likewise, immunohistochemistry (IHC) analysis of tumor sections generated in HCT116 cells presenting high PAR_2_ and PAR_4_ levels showed increased expression of p21 upon Pc(4-4) treatment; this was not seen in non-treated tumor sections. Similarly, increased levels of caspase-3 were observed, indicative of small tumors, as compared to the low caspase-3 levels seen in non-treated large tumor-tissue sections (Figure 7c,d).
3. Discussion
In this study, we demonstrated that PAR_2_ affects PAR_4_ oncogenic function. PAR_4_ requires PAR_2_ signaling for its pro-tumor roles. We showed that PAR_2_ co-localizes with PAR_4_ and acts as a single functional unit in promoting events in the β-catenin stabilization pathway and PAR-PH-Akt association. When Par2/f2rl1 is knocked down, the potent inhibition of β-catenin levels, β-catenin transcriptional activity, as reported by TOPflash, and increased nuclear DVL1 levels were obtained. Likewise, when Par2/f2rl1 was silenced, a powerful inhibition of PAR_2_ and PAR_4_-induced Matrigel invasion and cell migration/proliferation were seen. These observations are in contrast to the increased pro-tumor events induced by high PAR_2_ and PAR_4_ levels outlined above. TrPAR_2_, which is incapable of initiating cell signaling, greatly inhibited PAR_2_ and PAR_4_-induced β-catenin stabilization, TOPflash-reported transcriptional activity, Akt-PAR_4_ association, colony formation, and PAR_4_-induced stem-cell marker levels. These changes took place under conditions in which TrPAR_2_ effectively co-localized with PAR_4_. The importance of PAR_2_-induced signaling for PAR_4_ function was demonstrated Via the inhibition of PAR_2_ and PAR_4_-induced tumor generation in vivo. In contrast, potent tumor growth were generated by the inoculation of clones expressing both PAR_2_ and PAR_4_. To further understand the oncogenic mechanism of action of PAR_2_, we studied the impact of Pc(4-4), a PAR_2_ inhibitor directed to PAR_2_ and PAR_4_ PH-binding motifs. We demonstrated that upon Pc(4-4) treatment, increased levels of p53 were observed. In parallel, Pc(4-4) was shown to inhibit PAR-induced tumors [21]. In fact, Pc(4-4) is intended to be a drug in the fight against cancer. We are now in the process of measuring its half-life in blood and its efficient concentration, as well as its effect on physiological protease-activated PAR_2_ and PAR_4_.
We previously demonstrated that PAR_1_ and PAR_2_ act together as a single functional unit while promoting breast cancer growth [27], as also shown by others in different settings [26,27,28]. Whether the exposed internal ligand of the PAR_2_ sequence transactivates PAR_4_ or whether the PAR_2_–PAR_4_ heterodimer is formed by another route remains to be determined. The bioinformatics assignment of PAR_4_ as an oncogene was previously reported [15]. Along with this line of evidence, we have recently shown that PAR_4_ is a potent oncogene capable of inducing tumors in vivo [21]. However, publications offering opposing roles for PAR_4_ in cancer biology exist. Some describe PAR_4_ as playing an inhibitory role in cancer [42,43,44,45]. Still, an increasing number of publications point to a role of PAR_4_ as an oncogene [17,46,47,48,49,50,51,52,53,54].
PAR_4_ associates with PAR_1_ to form heterodimers following thrombin activation. Mapping the PAR_1_–PAR_4_ heterodimer interface showed it involving four residues in the TM4 (transmembrane 4) of both PAR_1_ and PAR_4_ [55]. The mechanistic basis for PAR_1_–PAR_4_ heterodimer generation indicates that in human platelets, PAR_1_ functions as a co-factor for thrombin activation of PAR_4_. Consequently, thrombin acts as a bivalent agonist of both receptors [56,57]. Whereas PAR_4_ is required for later stages of platelet function, PAR_1_ is essential for early stages in platelet activation [58].
The fact that PAR family members work in concert and are inter-dependent is interesting but not unique. The EGF receptor (EGFR)/erbB family, comprising EGFR/erbB1/Her1, erbB2/Her2, erbB3/Her3, and erbB4/Her4, provides another such example. erbB3 lacks kinase activity; thus, it cannot induce cell proliferation. Moreover, cells expressing a mutant form of Her2 incapable of binding EGF ligand exhibit a low rate of cell proliferation. Only cells expressing both erbB3 and mutant Her2 receptors show strong kinase activity and robust cell proliferation upon addition of an EGF ligand [58,59,60,61]. With regard to the PAR family, PAR_2_–PAR_4_ heterodimers are functional, and as demonstrated here, PAR_2_ acts as a dominant receptor, relative to PAR_4_ and PAR_1_ [27].
In fact, PARs transactivate EGFR, in this respect we have previously demonstrated that activation of PAR_4_ for example, directly induces EGFR tyrosine (Y) phosphorylation. Application of Pc(4-4) potently inhibits PAR-induced pY-EGFR [21].
Our findings reported here agree with previous work, demonstrating that the activation of PAR_2_ leads to the up-regulation of Bcl2L12 and the inhibition of p53 in lung cancer [58,62]. It is well known that p53 is a common denominator in the etiology of different sub-types of human cancers. Studies of p53 have contributed to our understanding of the cancerous process and provided insight into the development of tumor growth. Yet, one should keep in mind that signaling pathways linking p53-like cellular pathways should not be evaluated as an isolated element [63,64,65]. It is necessary to consider the intertwined networks into which p53-associated signaling is tightly linked. Mutant p53 is a bona fide useful biomarker impacting therapy programs such as those for EGFR [e.g., TKI (tyrosine kinase inhibitor)-EGFR] [37], the clinical regimen [38], and a useful biomarker for glioma [66]. A specific stop point in the cell-division cycle corresponds to the earliest effects of p53 expression and is seen in all mammalian cells. This arrest was mediated Via p53-induced expression of p21WAF1/CIP1, an inhibitor of cyclin-dependent kinases. The pivotal canonical activation of p53 leads to cell apoptosis [67]. The transcription of Bax, an apoptosis gene mediator, is directly activated by p53-binding sites in the gene regulatory region [68]. The multistage apoptotic process includes the release of mitochondrial proteins, like cytochrome c, and the activation of a cascade of cysteine proteases, like pro-caspase-9, which cleaves procaspase-3, leading to committed cell death [69,70]. p53 activity is often inhibited by numerous antagonistic mechanisms, the most noticeable of which is the activation of the Mdm2 E3 ligase [63,64,65].
Proteomic and transcriptomic analyses have convincingly demonstrated that the combined gene regulation of the wt p53 tumor suppressor and c-Myc oncogene is key in eradicating cancer stem cells [71]. Notably, p53 and c-Myc appear in many cancer networks. The robust increase in wt p53 levels seen upon treatment with Pc(4-4) highlight this compound as a powerful therapeutic compound. Similarly, the activation of wt p53 can be achieved by inhibitors of CKIα [72,73]. Small molecules that co-target kinase inhibitors (e.g., CKIα) in acute myeloid leukemia have been shown to induce p53 levels and inhibit c-Myc oncogene concomitantly [74]. Likewise, Pc(4-4) was found to be a potent inducer of wt p53 levels and its downstream effector p21, as well as caspase-3. In summary, we have demonstrated that in the absence of PAR_2_ signaling, PAR_4_ pro-tumor functions are significantly inhibited and Pc(4-4) acts as a potent anti-tumor agent Via a significant increase in wt p53 levels.
4. Materials and Methods
4.1. Animal Models
Animals used in the experiments were handled in accordance with the guidelines of the Hebrew University ethics committee (AAALAC standard). The animal ethics certificate number is MD-17368-5.
4.2. Cell Culture
HCT-116, HT29, RKO, LOVO, and HEK293 cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and grown in DMEM, supplemented with 1 mM L-glutamine, 50 µg/mL streptomycin, 50 U/mL penicillin (GIBCO-BRL, Gaithersburg, MD, USA) and 10% fetal calf serum (Biological Industries, Migdal HaEmek, Israel). These cell lines were checked for authentication by the service at the genomic center BCF biomedical core facilities, Haifa, Israel.
4.3. Matrigel Invasion
Blind-well chemotaxis chambers with 13 mm diameter filters were used. Matrigel (AcroBiosystems, Boston, Cambridge, MA, 02142, USA; AC-M082704) was applied per blind well (50 mg/filter) as described previously [24,26].
4.4. Scratch-Wound Healing
The scratch-wound healing assay was performed as described previously, with some modifications [75]. In brief, HT29 and HT29 shPar2 cells (3 × 10^6^/well) were seeded in 6-well plates. The cell medium was replaced with serum-free medium overnight, and equal wound areas were introduced into the monolayer. Cells were treated with 200 µM of the synthetic peptides SLIGKV or AYPGKF (GenScript; Piscataway, NJ, USA).
4.5. Clonogenic Assay
RKO (200) was seeded into 24-well plates, activated for both PAR_2_ and PAR_4_, and maintained until visible colonies were observed. The colonies were fixed with glutaraldehyde (6.0%, v/v; Sigma-Aldrich-Merck, Kiryat HaMada St 15, Jerusalem, Israel), stained with crystal violet (0.5%, w/v) and counted using a stereo microscope [76].
4.6. Plasmids and Reagents
pBJ-FLAG-Par4 (cat #53231), lrp6 (cat #27242), pCMV-VSV-G (cat #8454) and pCMV-dR8.2 dvpr (cat #8455) plasmids were purchased from Addgene (Watertown, MA, USA). Human PAR_2_ (Par2/f2rl1) plasmid was kindly provided by Dr. Morley D. Hollenberg (Faculty of Medicine, University of Calgary, Calgary, AB, Canada). flg-β-catenin was kindly provided by Dr. Ben-Neriah (Hebrew University, Jerusalem, Israel). Preparation of a TrPAR_2_ plasmid, encoding the protein lacking the cytoplasmic tail (i.e., lacking 150 residues) was carried out as previously described [25].
4.7. Cell Transfections and PAR Activation
Cells grown to 70–80% confluency were transfected with 0.5–1 μg of plasmid DNA using the PEI transfection reagent (Polysciences, Warrington, PA, USA). Cells were collected 48 h after transfection and protein lysates or RNA were prepared. Two hundred µM of the synthetic peptides SLIGKV or AYPGKF (GenScript; Piscataway, NJ, USA) were used to activate PAR_2_ and PAR_4_, respectively.
4.8. Small Hairpin (sh)-RNA Construct Preparation and Viral Particle Generation
To prepare sh-RNA of different genes and thereby knockdown gene expression, the desired sequence was successfully cloned into the plentilox3.7 (pLL3.7; #11795; Addgene, Watertown, MA, USA) lentiviral vector following the protocol provided by the Addgene website (cat #11795). The target sequences were sh-Par2i: 5′-GGAAGAAGCCTTATTGGTA-3′, sh-Par2ii: 5′-GCTCTTTGT AATGTGCTTA-3, scrambled sh-Par2i: AGAGAAGTTCGAAGGTATA-3′ and scrambled sh-Par2ii: 5′-TGCTGTGATAGTTAT TCGA-3′. For the generation of viral particles, HEK293 cells were transfected with three plasmid systems that encode packaging (CMVD R8.91), envelope (CMV-VSV-G) proteins, and cloned pLL3.7 vector using PEI as transfection reagent. The medium was replaced with fresh medium 24 h later. On day three after transfection, the medium was collected, and the viral particles were concentrated 10-fold by centrifuging for 2 h at 40,000 rpm.
4.9. Preparation of RKO Stable Clones Expressing wt PAR2/wtPAR4 and trPAR2/wtPAR4
RKO cells (0.5 × 10^6^) were infected with Par4 10 × viral particles along with Polybrene infection reagent. At 72 h post transduction, the cells were subjected to puromycin selection (0.5 µg/mL). While the control cells died, transduced cells with puromycin resistance grew and were collected. These cells were then infected with Par2 10 × virus particles along with Polybrene. Puromycin-resistant cells were collected and either used to isolate RNA or to prepare protein lysates.
4.10. Quantitative Real-Time (qRT) PCR and Reverse Transcriptase (RT) PCR
RNA was extracted from cells using the GenElute RNA kit (Sigma-Aldrich, St. Louis, MO, USA). To prepare cDNA, 1 µg of RNA was reverse-transcribed using reverse transcriptase (Promega, Madison, WI, USA (Quanta bio, Beverly, MA, USA)). qRT-PCR was performed using specific forward and reverse primers for each gene analyzed. In qRT-PCR amplifications, triplicates of the 6 ng cDNA template were used with 500 nM gene-specific primers and the 2 × PerfeCTa SYBR Green mix in an RG-3000A automated rotor-gene system (Corbett Research, Sydney, Australia).
4.11. Cell Lysate Preparations, Immunoprecipitation (IP), and Western Blot
To prepare protein-cell lysates for IP, cells were solubilized in CelLytic M buffer (Sigma-Aldrich, Saint Louis, MO, USA). Protein-cell lysates were prepared as previously described [12,14]. Western blot analysis was performed as in previous studies [10,11,12,13,14]. In this study, experimental cell-protein extraction and lysis were carried out using RIPA (Radio-Immunoprecipitation Assay) buffer supplemented with PMSF and a protein inhibitor. Subsequently, protein concentrations were determined, and the lysates were heated with a loading buffer at 100 °C for 10 min. Thirty micrograms of proteins from each sample were loaded onto 10%. SDS-PAGE gels, transferred to PVDF membranes, blocked with 5% BSA, and then exposed to the relevant antibody. Western blots (wet Western blots) were performed. Membranes (PVDF, Thermofisher, Waltham, MA, USA; TS-88518) were blocked and probed with the appropriate primary antibodies. These are anti-FLAG (F3165; Sigma-Aldrich, Saint Louis, MO, USA), anti-β-actin (A5441; Sigma-Aldrich, Saint Louis, MO, USA), anti-β-catenin (C2206), anti-PAR_2_ (AB180953; Abcam, Cambridge, UK and SC-13,504 Santa Cruz Biotechnology, Dallas, TX, USA), anti-PAR_4_ (AB5787; Abcam (Cambridge, UK): SC-13504 Santa Cruz Biotechnology (Dallas, TX, USA)), anti-HA (901503; Biolegend, San Diego, CA, USA), anti-LRP6 (BS-7007R, Bioss Antibodies, Woburn, MA, USA), anti-phospho-LRP6 (Cell Signaling Technology, Danvers, MA, USA), anti-AKT (AB8805; Abcam, Cambridge, UK), anti-p53 (AB17990; Abcam, Cambridge, UK), anti-p21 (AB109520; Abcam, Cambridge, UK), and anti-GAPDH (AB9485; Abcam, Cambridge, UK). These antibodies were suspended in 3% BSA (#A5611, Sigma-Aldrich, Head office Kanagawa, Japan) in 10 mM Tris-HCl (T1503; Sigma-Aldrich, Saint Louis, MO, USA), pH 7.5, 100 mM NaCl, and 0.1% Tween-20 (P9416; Sigma-Aldrich, Saint Louis, MO, USA). After extensive washes, blots were incubated with secondary antibodies conjugated to horseradish peroxidase (HRP) anti-mouse (#ab6728; Abcam, Cambridge, UK) or anti-Rabbit (Abcam, Cambridge, UK; #ab6721). Immunoreactive bands were detected by the enhanced chemiluminescence (ECL) reagent (Pierce, Rockford, IL, USA).
4.12. TOPflash Luciferase Reporter
HEK 293 cells (0.2 × 10^6^) were seeded in 6-well plates and incubated overnight at 37 °C. The cells were transfected with desired plasmids (wtPar2, trPar2 and wtPar4) and TOPflash components, as previously described [12].
4.13. Ectopic Tumor Xenograft Mouse Model
RKO cells (wt and stable clones of RKOtrPAR_2_, RKOwtPAR_2_ + wtPAR_4_ and RKOtrPAR_2_ + wtPAR_4_) were starved overnight, and treated the next day with the SLIGKV or AYPGKF peptides (200 µM each) for 4 h. The cells (1 × 10^6^) were injected subcutaneously into the right flank of groups of five six–eight-week-old Hsd: Athymic NudeFoxn1nu mice (nude mice). The mice were terminated when the tumor volumes reached the volume stipulated by the Animal Committee approval.
4.14. Immunohistochemistry (IHC)
Tumor tissue-derived paraffin-embedded slides were used for IHC, as previously described [23,25].
4.15. Pc(4-4)
Pc(4-4) is a cyclic peptide directed toward PAR_2_ and PAR_4_ PH-binding motifs. More detailed description appears in [21].
5. Conclusions
We have demonstrated that PAR_2_ is dominant over PAR_4_ in colon cancer development. The shRNA silencing of Par2/f2rl1inhibits PAR_2_ and PAR_4_-induced events in the β-catenin stabilization pathway, and inhibits invasion and migration. Similarly, truncated PAR_2_ (TrPAR_2_), devoid of the cytoplasmic tail of PAR_2_, inhibits PAR_2_ and PAR_4_-induced β-catenin stabilization, PAR_4_-Akt association, stem-cell marker expression and colony formation. TrPAR_2_ inhibits xenograft PAR_2_ and PAR_4_-induced tumor growth in vivo. Pc(4-4), a compound directed to the PAR_2_ PH-binding domain, inhibits PAR_2_ oncogenic activity Via an increase in p53 levels.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Dorsam R.T. Gutkind J.S. G-protein-coupled receptors and cancer Nat. Rev. Cancer 20077799410.1038/nrc 206917251915 · doi ↗ · pubmed ↗
- 2Feigin M.E. Harnessing the genome for characterization of G-protein coupled receptors in cancer pathogenesis FEBS J.20132804729473810.1111/febs.1247323927072 PMC 4283816 · doi ↗ · pubmed ↗
- 3Lappano R. Maggiolini M. G protein-coupled receptors: Novel targets for drug discovery in cancer Nat. Rev. Drug Discov.201110476010.1038/nrd 332021193867 · doi ↗ · pubmed ↗
- 4Siegel R.L. Giaquinto A.N. Jemal A.C.A. Cancer statistics, 2024 Cancer J. Clin.202474124910.3322/caac.2182038230766 · doi ↗ · pubmed ↗
- 5Sonkin D. Thomas A. Teicher B.A. Cancer treatments: Past, present, and future Cancer Genet.2024286182410.1016/j.cancergen.2024.06.00238909530 PMC 11338712 · doi ↗ · pubmed ↗
- 6Nusse R. Clevers H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities Cell 201716998599910.1016/j.cell.2017.05.01628575679 · doi ↗ · pubmed ↗
- 7Koo B.-K. Spit M. Jordens I. Low T.Y. Stange D.E. Van De Wetering M. Van Es J.H. Mohammed S. Heck A.J.R. Maurice M.M. Tumour suppressor RNF 43 is a stem-cell E 3 ligase that induces endocytosis of Wnt receptors Nature 201248866566910.1038/nature 1130822895187 · doi ↗ · pubmed ↗
- 8Hao H.-X. Xie Y. Zhang Y. Charlat O. Oster E. Avello M. Lei H. Mickanin C. Liu D. Ruffner H. ZNRF 3 promotes Wnt receptor turnover in an R-spondin-sensitive manner Nature 201248519520010.1038/nature 1101922575959 · doi ↗ · pubmed ↗