Phenotypical Characterization of C9ALS Patients from the Emilia Romagna Registry of ALS: A Retrospective Case–Control Study
Andrea Ghezzi, Giulia Gianferrari, Elisa Baldassarri, Elisabetta Zucchi, Ilaria Martinelli, Veria Vacchiano, Luigi Bonan, Lucia Zinno, Andi Nuredini, Elena Canali, Matteo Gizzi, Emilio Terlizzi, Doriana Medici, Elisabetta Sette, Marco Currò Dossi, Simonetta Morresi

TL;DR
This study compares the clinical features of ALS patients with and without C9ORF72 mutations, finding faster disease progression and sex-specific differences in C9ALS patients.
Contribution
The study provides new insights into the phenotypic differences and sex-specific characteristics of C9ALS patients compared to nmALS patients.
Findings
C9ALS patients showed faster disease progression and shorter times to gastrostomy and ventilation.
Female C9ALS patients had more severe bulbar and upper motor neuron involvement.
Cognitive symptoms were more common in C9ALS and were an independent prognostic factor.
Abstract
Background/Objectives: C9ORF72 expansion is associated with significant phenotypic heterogeneity. This study aimed to characterize the clinical features of C9ALS patients from the Emilia Romagna ALS registry (ERRALS) and compare them with non-mutated ALS (nmALS) patients matched for sex, age at onset, and diagnostic delay, sourced from the same register. Methods: In total, 67 C9ALS patients were compared to 201 nmALS. Clinical data, phenotype, and prognostic factors were analyzed in the two groups and within the C9ALS group after stratification by sex. Results: C9ALS patients displayed a higher disease progression rate and shorter times to gastrostomy and invasive ventilation, despite no differences in overall survival. Female C9ALS had a more severe bulbar and upper motor neuron involvement compared to males. Cognitive and behavioral symptoms were more common in the C9ALS group, and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
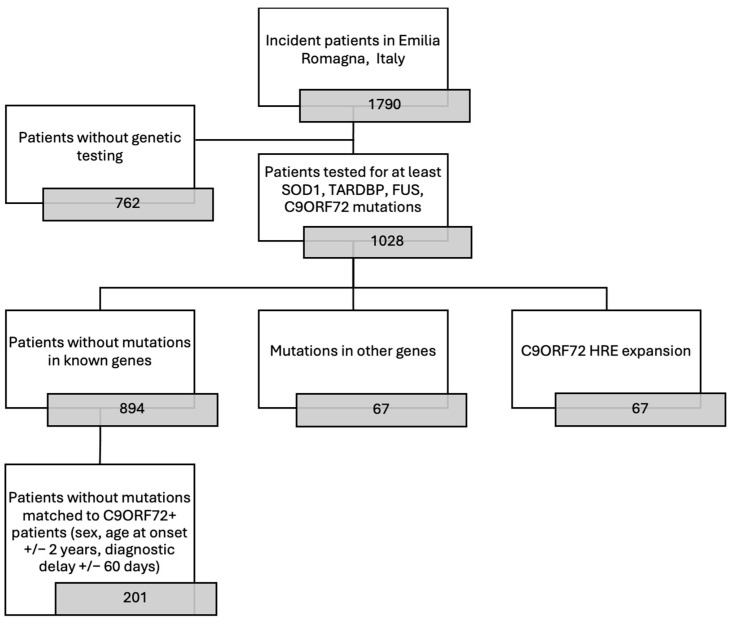 Figure 1
Figure 1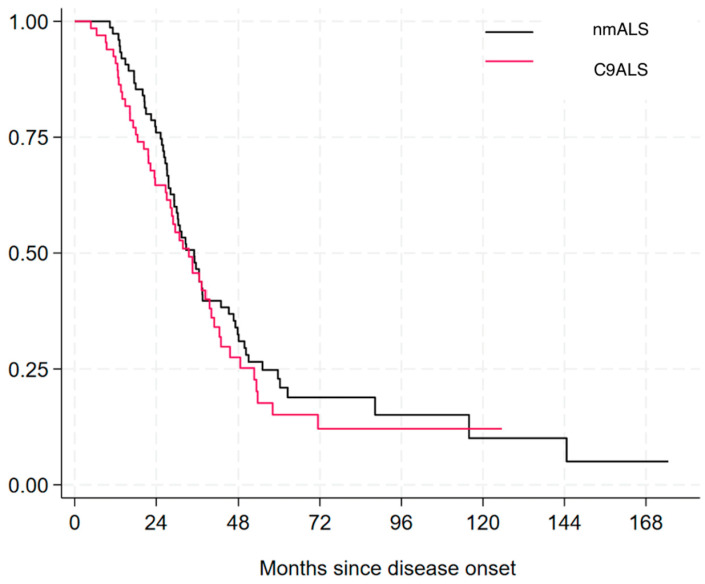 Figure 2
Figure 2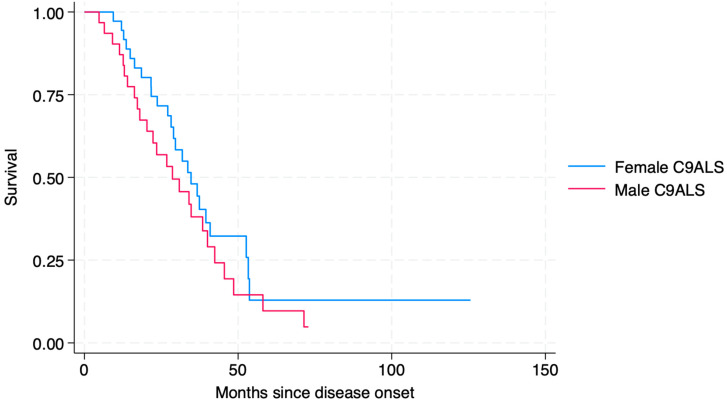 Figure 3
Figure 3- —Emilia Romagna Regional Health Authority
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAmyotrophic Lateral Sclerosis Research · Biochemical Acid Research Studies · Neurogenetic and Muscular Disorders Research
1. Introduction
Hexanucleotide (G4C2)n repeat expansions (HREs) in the non-coding region of chromosome 9 open reading frame 72 (C9ORF72) are responsible for 30–50% of familial amyotrophic lateral sclerosis (fALS) and 7–10% of sporadic ALS (sALS) cases [1], as well as 5–10% of frontotemporal dementia (FTD) cases [2].
In healthy individuals, C9ORF72 typically contains eleven or fewer hexanucleotide repeats, whereas ALS patients may present with hundreds to thousands of repeats. Although a clear pathological threshold has yet to be defined, most studies use a cutoff of 30 repeats as a reference [3].
C9ORF72 mutations are inherited in an autosomal dominant pattern, with incomplete and age-dependent penetrance: the likelihood of developing symptoms rises from 50% at 58 years of age to 99.5% by age 83 [4].
Clinically, the C9ORF72-associated phenotype is highly variable. ALS associated with C9ORF72 HRE (C9ALS) is more frequently characterized by a bulbar onset manifesting as dysphagia and dysarthria, with a higher incidence compared to ALS cases overall (30–40% vs. 25–30%, respectively) [5]. Cognitive involvement is also more common in C9ALS patients, with 20% of patients showing cognitive impairment, 10% exhibiting behavioral symptoms, and 20% meeting the criteria for the diagnosis of FTD [5].
Psychiatric symptoms are also prominent in patients with C9ORF72 HRE, with 20–60% manifesting psychotic signs such as delusions and hallucinations, but also obsessive/compulsive disorder and catatonia [6].
Parkinsonism is observed in up to 60% of patients and can sometimes be the initial symptom, with motor neuron or cognitive deficits emerging years later [7].
Notably, around 5% of patients with a clinical presentation resembling Huntington’s disease but without the CAG expansion in HTT gene harbor an expansion in C9ORF72 [7].
Despite extensive research, the pathomechanism behind the clinical heterogeneity of C9ORF72-associated diseases remains poorly understood. Some studies have attempted to correlate disease severity and age of onset with HRE length, with inconsistent results [8]. Somatic mosaicism, where the number of HRE varies among different tissues within the same individual, further complicates the genetic landscape. For example, the repeat size in blood can differ from that in the brain [9], which has implications for both genetic testing and the resulting disease phenotype.
To better understand the role of genetics in C9ALS, given that the spatial–temporal combination of motor and cognitive events leading to ALS onset and progression is influenced by factors such as age, sex [10], and gene variants [11], we conducted a retrospective case–control study. We thoroughly characterized phenotypically the C9ALS patients from the Italian Emilia-Romagna region’s ALS (ERRALS) registry and compared them with age- and sex-matched non-mutated ALS patients from the same registry.
2. Methods
2.1. Patients’ Data Collection
A total of 67 patients from the ERRALS registry [12], with a diagnosis of ALS according to El Escorial revised criteria [13] between 2009 and 2024, carrying an HRE in C9ORF72 gene, were included and matched to ALS patients from the same register with no mutation in the four major ALS-related genes (SOD1, FUS, TARDBP, and C9ORF72), in order to avoid the potential effect of the other mutations on disease clinical features and progression, for a total of 201 non-mutated ALS patients (nmALS). Matched nmALS patients were selected among patients from the same registry of the same sex, age at onset (+/− 3 years), and diagnostic delay (i.e., the time between the onset of symptoms and the diagnosis (+/− 90 days)). In the case of multiple matches, nmALS patients diagnosed in the same period as C9ALS were chosen, as described in the flowchart (Figure 1). C9ORF72 status was determined by repeat primed PCR, using the AmplideX^®^ PCR/CE C9orf72 Kit (Asuragen Inc., Austin, TX, USA), which allows to precisely quantify HRE’s lengths up to approximately 145 repeats, above which the expansion is accurately detected but not precisely quantified. The other mutations were identified using a panel that included up to 78 ALS-associated genes, as previously described [14,15].
Clinical data from all patients were collected at diagnosis and over disease course, as previously reported [12], including demographics, age at onset and diagnosis, site and time of onset, phenotype (classified as classic, bulbar, upper motor neuron (UMN) predominant, flail arm, flail leg, respiratory) [16], clinical signs such as spasticity, pathological reflexes, clonus, cramps, cognitive and/or behavioral involvement according to Strong’s criteria [17], comorbidities, drug history (including Riluzole) [18], familial history of neurodegenerative diseases (ALS, FTD, Parkinson’s disease, Alzheimer’s disease), weight, body mass index (BMI) and forced vital capacity (FVC) assessed by spirometry, time to generalization [19], disease progression using ALSFRS-r scale and disease progression rate, measured considering ALSFRS-r at diagnosis and at the last follow-up visit [20]. Data regarding the need for non-invasive ventilation (NIV), invasive ventilation (IV), enteral feeding through percutaneous endoscopic gastrostomy (PEG), and date, place, and cause of death were also gathered [21].
The disease progression rate at diagnosis was calculated as follows:
Absolute weight loss at diagnosis was defined as the difference in kilograms between the body weight during healthy status and the time of diagnosis, while relative weight loss was calculated as the percentage of the healthy weight that was lost at the time of diagnosis.
Data regarding single clinical manifestations and compound UMN and lower motor neuron (LMN) scores were also collected and quantified by the Penn Upper Motor Neuron Score (PUNMS) [22] and Devine Lower Motor Neuron Score (DLMNS) [23].
2.2. Statistical Analysis
We assessed differences across ALS patients’ groups by using a t test, ANOVA, or Chi-square tests as appropriate.
Regression analyses were conducted to evaluate the influence of clinical features on the disease progression rate.
Survival analysis was conducted using Kaplan–Meier curves, and the Log-rank test was applied for univariate analyses, while multivariate analyses were performed using the Cox regression model (by stepwise backward method).
Data analysis was performed using the STATA statistical package 15 (StataCorp. 2017. College Station, TX, USA: StataCorp LLC).
3. Results
Out of over 1028 patients included in the Emilia Romagna Register for ALS and tested for at least the four main genes related to ALS, 67 (6.52%) showed C9ORF72 expansion, whereas 894 (86.96%) did not show any further mutation in SOD1, FUS, or TARDBP genes.
The 201 patients that best matched the 67 C9ORF72 carriers were selected first based on diagnostic delay (±90 days) and then on sex and age at onset (±3 years).
Among the 67 C9ALS patients, 7 (10.4%) had an HRE length shorter than 145 repeats, which could be precisely quantified. In contrast, in the remaining 60 patients (89.6%), the expansion exceeded 145 repeats and could not be precisely determined (see Methods Section).
Within the nmALS cohort, some patients carried variants of uncertain significance (VUS) in the following genes: TBK1 (n = 1), CHMP2B (n = 1), DCTN1 (n = 1), KIF5A (n = 1), MAPT (n = 3), FIG4 (n = 1), and SQSTM1 (n = 1).
3.1. Demographic and Clinical Features of ALS Patients with and Without C9ORF72 Expansion
General features of the two groups are displayed in Table 1. The male/female ratio was 0.86 (31 males and 36 females among C9ALS and 93 males and 108 females among nmALS).
Family history for ALS and other neurodegenerative diseases was significantly more frequent in C9ALS patients as well as familiarity for psychiatric diseases (Table 1). Both first- and second-degree relatives for neurodegenerative diseases were more frequent in the C9ALS patients.
There were no differences in weight, BMI, and absolute and relative weight loss at diagnosis. Despite no significant difference being found in the ALSFRS-r total score at diagnosis, the bulbar sub-score was significantly lower in the C9ALS group (mean score 9.45 ± 2.42 in C9ALS group vs. 10.56 ± 2.24 in nmALS group, p = 0.050). FVC at diagnosis was slightly lower for the C9ALS group (89.18 ± 19.34 vs. 95.65 ± 21.86, p = 0.057). The time to gastrostomy and to IV was significantly shorter in C9ALS patients. Coherently, the progression rate was slower in nmALS both at diagnosis and at the last observation.
Among C9ALS patients, there were no phenotypic differences between those with more than 145 repeats and those with fewer.
3.2. Phenotype of ALS Patients with and Without C9ORF72 Expansion
Bulbar onset and phenotype were more frequent in the C9ALS group compared to the nmALS group (Table 2).
The compound clinical scores of upper and lower motor neuron involvements such as the DLMNS and the PUMNS did not show any difference between the two groups.
When looking at clinical symptoms, no significant difference was found in all of the clinical symptoms investigated, except for cramps, which were significantly less frequent in the C9ALS group (Table 2).
3.3. Cognitive Involvement of ALS Patients with and Without C9ORF72 Expansion
Cognitive and behavioral involvement was significantly more frequent in C9ALS patients compared to the nmALS group (Table 3). Following Strong’s criteria, 33.33% C9ALS patients could be classified with ALS-associated behavioral impairment (biALS) and 29.82% with ALS-associated cognitive impairment (ciALS), and 27.27% met the diagnostic criteria for FTD.
3.4. Comorbidities of ALS Patients with and Without C9ORF72 Expansion
Autoimmune diseases and dyslipidemia were significantly more frequent in the C9ALS group compared to nmALS patients (13.43% vs. 3.48%, p = 0.003 and 38.30% vs. 16.40%, p = 0.001, respectively). No significant difference was found in other comorbidities between C9ALS and nmALS patients except for depression, which was more frequent in the second group (21.21 vs. 39.74, p = 0.017) (Table 4).
3.5. Progression Rate and Survival of ALS Patients with and Without C9ORF72 Expansion
Regression analysis confirmed that the presence of C9ORF72 expansion (Coef: 0.45, 95% CI: 0.04 to 0.87, p = 0.033), as well as a younger age at onset (Coef: −0.03, 95% CI: −0.05 to −0.007, p = 0.008), led to a faster disease progression rate.
No difference was found in overall survival between the two groups (33.48 ± 18.85 vs. 39.42 ± 28.12, p = 0.199) (Figure 2).
Univariate Cox regression analysis and multivariate analysis for patients with C9ALS are shown in Table 5. In C9ALS patients, the multivariate analysis of survival showed that independent prognostic factors for tracheostomy-free survival were diagnostic delay (HR = 0.92, 95% CI 0.86–0.98, p = 0.014), disease progression rate at diagnosis (HR = 1.65, 95% CI 1.10–2.47, p = 0.016), and presence of cognitive involvement (HR = 7.70 95% CI 3.12–19.02, p < 0.001).
3.6. Sex-Related Differences in C9ALS Patients
When comparing clinical differences between sexes within the nmALS and the C9ALS groups (Table 6), no significant sex-related differences were found in the main clinical features, except for diagnostic delay, which was significantly shorter in male nmALS patients.
A higher disease progression rate at diagnosis was found for C9ALS males, despite not reaching clinical significance; this trend, albeit not significant, was confirmed also for time from symptoms onset to tracheostomy and death.
The site of onset was more frequently bulbar in the female population in both groups, and the ALSFRS-r bulbar subscale score was significantly higher in male C9ALS patients (10.16 ± 2.08 vs. 8.87 ± 2.56, p = 0.047).
While no differences were found in DLMNS, the PUMNS resulted in being significantly higher in females compared to males (9.92 ± 5.75 vs. 5.53 ± 5.12, p = 0.004).
Finally, when considering comorbidities, autoimmune diseases and psychosis were more frequent in female C9ALS patients, despite not reaching statistical significance
Despite a tendency towards a higher disease progression rate, shorter time to IV, or death in male C9ALS patients, no statistically significant difference in survival was found between men and women (Figure 3).
When analyzing prognostic factors in the C9ALS population stratified by sex, we found that cognitive changes (HR 3.87, 95% CI 1.12–13.34, p = 0.032), disease progression rate at diagnosis (HR 5.06, 95% CI 2.05–12.48, p < 0.001), and concomitant psychosis (HR 8.23, 95% CI 1.12–60.75, p = 0.039) were independent prognostic factors in women, whereas weight loss at diagnosis was the only independent prognostic variable in men (HR 1.16, 95% CI 1.02–1.33, p = 0.023) (Supplementary Table S1).
4. Discussion
In this study, we explore the phenotypic heterogeneity of ALS associated with the C9ORF72 mutation [5], comparing a population-based cohort of C9ALS patients with nmALS patients matched for sex, age, and diagnostic delay, sourced from the same population-based register.
As expected [24], we observed that both bulbar onset and bulbar phenotype were more frequent in the C9ALS cohort compared to the nmALS group. This was further corroborated by a significantly lower ALSFRS-r bulbar subscale score at diagnosis in the C9ALS group. However, unlike previous studies [25], bulbar onset in our C9ALS cohort was not associated with shorter survival. Interestingly, we identified a sex-related difference in bulbar involvement at diagnosis, with the ALSFRS-r bulbar subscale being lower in women, showing consistency with prior findings [11,25,26]. The mechanism behind the more frequent bulbar involvement in C9ALS is poorly understood, but some studies have identified multiple molecular subtypes in ALS patients which differed between bulbar and spinal onset patients, suggesting that different motor neurons could be susceptible to different pathological mechanisms [27]. For example, bulbar motor neurons may be more vulnerable to the pathological mechanisms that characterize C9ALS but are absent in nmALS, such as RNA foci formation, DPR accumulation, and C9orf72 loss of function [1].
We analyzed UMN and LMN involvement using two compound scores, PUMNS and DLMNS, respectively, in both C9ALS and nmALS cohorts. While no significant differences in global UMN and LMN scores were observed between the two groups, the stratification of C9ALS patients by sex revealed significantly higher PUMNS scores in females, indicating more prominent UMN involvement in this subgroup. No sex-related differences in PUMNS were found among nmALS patients, suggesting that increased UMN involvement may specifically affect female C9ALS patients. These findings point to the existence of sex-related differences in C9ALS pathology.
Family history of ALS was significantly more prevalent in C9ALS patients (up to 43.93%) compared to nmALS patients, consistent with C9ORF72 being the most common genetic cause of familial ALS [1]. Furthermore, the prevalence of family history for all neurodegenerative diseases in the C9ALS cohort reached 81.82%, underscoring the broader role of C9ORF72 in neurodegeneration [8]. Supporting this hypothesis, evidence suggests that C9orf72 regulates microglial amyloid clearance [28], potentially linking its dysfunction to broader neurodegenerative processes.
Indeed, the clinical spectrum of C9ORF72 HRE includes movement disorders and FTD [7]. It is plausible that, in some cases, Parkinsonism and dementia represent manifestations of C9ORF72 pathology without overt motor neuron involvement. In our cohort, only a few patients presented with Parkinsonism or psychosis, with no significant differences between the two groups. Depression, however, was significantly less frequent in the C9ALS cohort, possibly reflecting a lack of insight associated with cognitive involvement [29]. A family history of psychiatric diseases was significantly more frequent in C9ALS patients, supporting the role of C9ORF72 in psychiatric disease development [30].
Interestingly, psychiatric disorders were more common in females compared to males, suggesting a potential role for additional factors, such as hormonal influences, in the neuropsychiatric manifestations of C9ALS. Psychiatric involvement also emerged as an independent prognostic variable in female C9ALS patients. Although the association between C9ALS and psychiatric symptoms is well established [30], their impact on prognosis remains underexplored. The association between worst progression and psychiatric involvement, which is frequently seen in FTD [31], may simply reflect the poorer outcomes of the association of FTD in C9ALS patients [32].
Moreover, in our study, we observed a significantly higher incidence of autoimmune diseases in the C9ALS cohort, a result in line with the biological data on autoimmune phenotype in C9ORF72 knockout mouse models [33,34]. Despite the established relation between loss of function of C9orf72 and autoimmunity, only one prior study has reported an increased incidence of autoimmune diseases in carriers of the C9ORF72 HRE [35]. In our cohort, autoimmune diseases were more prevalent in female C9ALS patients than in males, though this difference did not reach statistical significance. While this observation might simply reflect the generally higher incidence of autoimmune diseases in females [36], emerging evidence highlights a critical role of C9ORF72 in immunity [37,38]. Therefore, a potential contribution of C9ORF72 HRE to autoimmunity in females cannot be ruled out.
Furthermore, the higher prevalence of dyslipidemia observed in female C9ALS patients compared to males suggests a complex interplay between genetic and hormonal factors in modulating metabolism [39]. The relationship between dyslipidemia and systemic inflammation is well documented [40] and may reflect metabolic dysfunction driven by the C9ORF72 mutation. This aligns with the mutation’s known involvement in immune regulation and inflammatory processes [41]. These findings, together with the increased incidence of autoimmune diseases, support the hypothesis of a broader role for C9orf72 in regulating metabolic and immune homeostasis [42]. A deeper understanding of the role of C9orf72 in metabolism and immunity could help identify specific molecular signatures for C9ALS, potentially enabling personalized treatment strategies. This approach has been recently suggested in studies investigating ALS-specific signatures in blood and CSF [43,44].
C9ALS patients are typically reported to have shorter survival, but prior studies on this topic have yielded conflicting results [25,45]. In our cohort, we found a significantly higher disease progression rate in C9ALS patients at both diagnosis and last observation. These patients also exhibited significantly shorter times to PEG and tracheostomy, consistent with the earlier and more frequent bulbar involvement in this cohort. Some studies have attempted to correlate the HRE length with disease progression and survival, but the findings remain inconclusive [8,45,46].
Unfortunately, since genetic testing was performed in a clinical setting, we do not have the exact number of C9ORF72 repeat expansions when the expansion exceeds a threshold of 145 repeats, which is widely recognized as above the pathological cutoff [7]. Precise sizing for very large expansions in fact requires complementary methods such as Southern blot analysis, which is not commonly used in clinical practice.
As a result, for most patients, data on the precise length of the HRE are unavailable, representing a significant limitation of our study. When analyzing potential correlations between the HRE length and key clinical variables, we found no significant differences between C9ALS patients with >145 and < 145 repeats. However, this could be due to the limited size of our sample.
Although not reaching statistical significance, our data showed reduced respiratory function at diagnosis in C9ALS patients. This observation might suggest the early subclinical involvement of respiratory muscles, particularly the phrenic motor neurons, in the initial stages of the disease [47]. Supporting this, we observed a significantly shorter time from diagnosis to tracheostomy in C9ALS patients. Previous studies have also reported increased vulnerability of phrenic MNs in C9ORF72-mutated iPSC models [47].
We did not find, however, a significant difference in overall survival, probably because of the small size of our cohort.
Multivariate analysis in our cohort of C9ALS patients highlighted the disease progression rate at diagnosis, time to diagnosis, and cognitive involvement classified as ALSci, as independent prognostic variables, consistent with previous findings [32].
Finally, we confirm that females with C9ORF72 expansions are more likely to present with a bulbar phenotype independent of age and are more prone to developing neuropsychiatric symptoms [25,31]. Sex differences in C9ALS patients’ disease progression were also evident, with males generally experiencing a more aggressive course and shorter survival times [10]. This is in line with the known hormonal influences, in particular the neuroprotective effects of estrogen, which may contribute to attenuating disease progression in females [48]. While the pathological hallmark of C9ORF72-associated diseases—TDP-43 proteinopathy—appears similar in males and females, subtle differences in its regional distribution or severity might exist and contribute to the reported differences. Further studies will be needed to address those discrepancies [49].
Further research to document sex differences in C9ORF72 expansions could have important implications for developing tailored therapeutic strategies in the future.
5. Conclusions
This study highlights the phenotypic heterogeneity of C9ORF72-associated ALS, revealing significant clinical and sex-related differences compared to non-mutated ALS. A family history of ALS and neurodegenerative diseases was significantly more common in C9ALS, supporting its broad role in neurodegeneration. Bulbar involvement was more frequent and severe in C9ALS, with women showing greater upper motor neuron involvement and more pronounced bulbar dysfunction. Psychiatric symptoms, particularly in females, emerged as a distinct feature and independent prognostic factor, potentially influenced by hormonal and genetic factors.
C9ALS patients demonstrated faster disease progression and an earlier need for interventions, though overall survival did not differ from nmALS, possibly due to the limited cohort size.
Sex differences in disease progression, psychiatric and autoimmune involvement, and bulbar phenotypes emphasize the importance of tailoring therapeutic strategies. Future research should focus on the interplay between genetic, immune, and sex-specific factors to better understand and manage C9ALS.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Masrori P. Van Damme P. Amyotrophic lateral sclerosis: A clinical review Eur. J. Neurol.2020271918192910.1111/ene.1439332526057 PMC 7540334 · doi ↗ · pubmed ↗
- 2Greaves C.V. Rohrer J.D. An update on genetic frontotemporal dementia J. Neurol.20192662075208610.1007/s 00415-019-09363-431119452 PMC 6647117 · doi ↗ · pubmed ↗
- 3Balendra R. Isaacs A.M. C 9orf 72-mediated ALS and FTD: Multiple pathways to disease Nat. Rev. Neurol.20181454455810.1038/s 41582-018-0047-230120348 PMC 6417666 · doi ↗ · pubmed ↗
- 4Murphy N.A. Arthur K.C. Tienari P.J. Houlden H. ChiòA. Traynor B.J. Age-related penetrance of the C 9orf 72 repeat expansion Sci. Rep.20177211610.1038/s 41598-017-02364-128522837 PMC 5437033 · doi ↗ · pubmed ↗
- 5Zampatti S. Peconi C. Campopiano R. Gambardella S. Caltagirone C. Giardina E. C 9orf 72-Related Neurodegenerative Diseases: From Clinical Diagnosis to Therapeutic Strategies Front. Aging Neurosci.20221490712210.3389/fnagi.2022.90712235754952 PMC 9226392 · doi ↗ · pubmed ↗
- 6Benussi A. Premi E. Gazzina S. Brattini C. Bonomi E. Alberici A. Jiskoot L. van Swieten J.C. Sanchez-Valle R. Moreno F. Genetic FTD Initiative (GENFI). Progression of Behavioral Disturbances and Neuropsychiatric Symptoms in Patients with Genetic Frontotemporal Dementia JAMA Netw. Open 20214 e 2030194 Erratum in: JAMA Netw. Open 2021, 4, e 21766410.1001/jamanetworkopen.2020.3019433404617 PMC 7788468 · doi ↗ · pubmed ↗
- 7Van der Ende E.L. Jackson J.L. White A. Seelaar H. van Blitterswijk M. Van Swieten J.C. Unravelling the clinical spectrum and the role of repeat length in C 9ORF 72 repeat expansions J. Neurol. Neurosurg. Psychiatry 20219250250910.1136/jnnp-2020-32537733452054 PMC 8053328 · doi ↗ · pubmed ↗
- 8Smeyers J. Banchi E.G. Latouche M. C 9ORF 72: What It Is, What It Does, and Why It Matters Front. Cell Neurosci.20211566144710.3389/fncel.2021.66144734025358 PMC 8131521 · doi ↗ · pubmed ↗