Inherited Dyslipidemic Splenomegaly: A Genetic Macrophage Storage Disorder Caused by Disruptive Apolipoprotein E (APOE) Variants
Elise A. Ferreira, Machteld M. Oud, Saskia N. van der Crabben, Miranda Versloot, Susan M. I. Goorden, Clara D. M. van Karnebeek, Jeffrey Kroon, Mirjam Langeveld

TL;DR
A rare inherited disorder caused by APOE gene variants leads to enlarged spleen and lipid storage in macrophages, mimicking lysosomal storage diseases.
Contribution
Identifies a novel inherited metabolic disorder caused by disruptive APOE variants with macrophage storage and splenomegaly.
Findings
APOE variants (ε1/ε1) cause a macrophage storage disorder resembling lysosomal storage diseases.
Patients with these variants show increased lipid content in monocytes and foam cell formation in tissues like the spleen.
Lipid-lowering therapy and dietary changes can partially reverse symptoms, while splenectomy is contraindicated.
Abstract
Background: Persistent splenomegaly, often an incidental finding, can originate from a number of inherited metabolic disorders (IMDs). Variants of APOE are primarily known as risk factors in terms of cardiovascular disease; however, severe dysfunction of APOE can result in a disease phenotype with considerable overlap with lysosomal storage disorders (LSDs), including splenomegaly and gross elevation of N-palmitoyl-O-phosphocholine-serine (PPCS). Methods: A case study (deep phenotyping, genetic and FACS analysis) and literature study was conducted. Results: The index patient, with a family history of early-onset cardiovascular disease, presented with splenic infarctions in a grossly enlarged spleen. The identified genetic cause was homozygosity for two APOE variants (c.604C>T, p.(Arg202Cys) and c.512G>A, p.(Gly171Asp); ε1/ε1), resulting in a macrophage storage phenotype resembling an…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
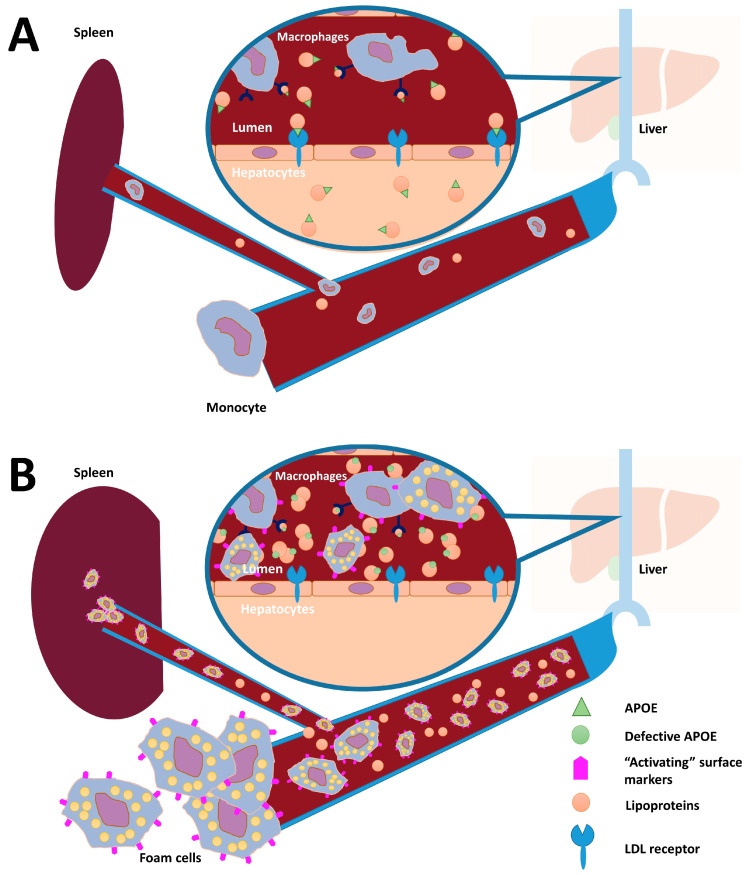 Figure 1
Figure 1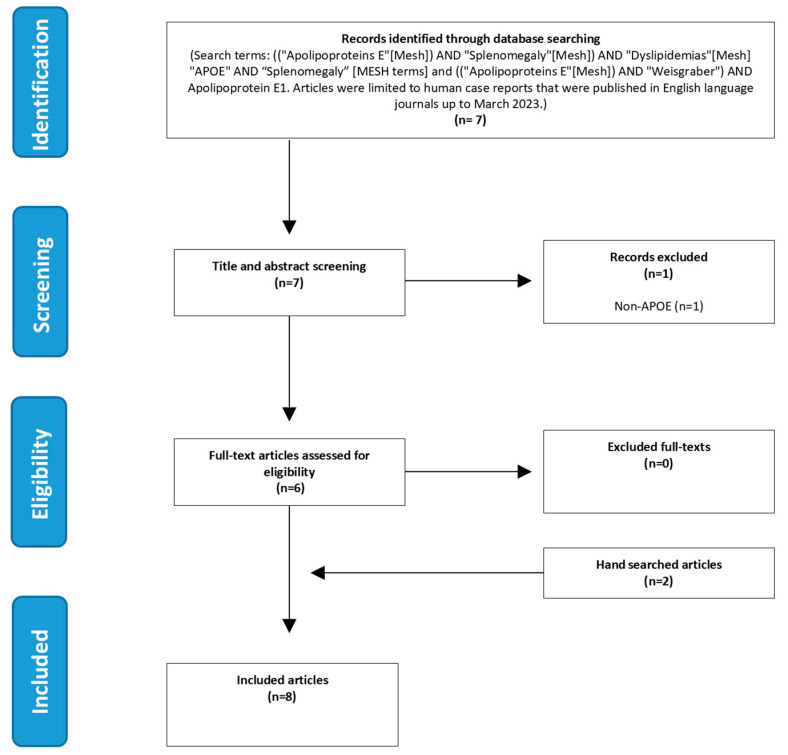 Figure 2
Figure 2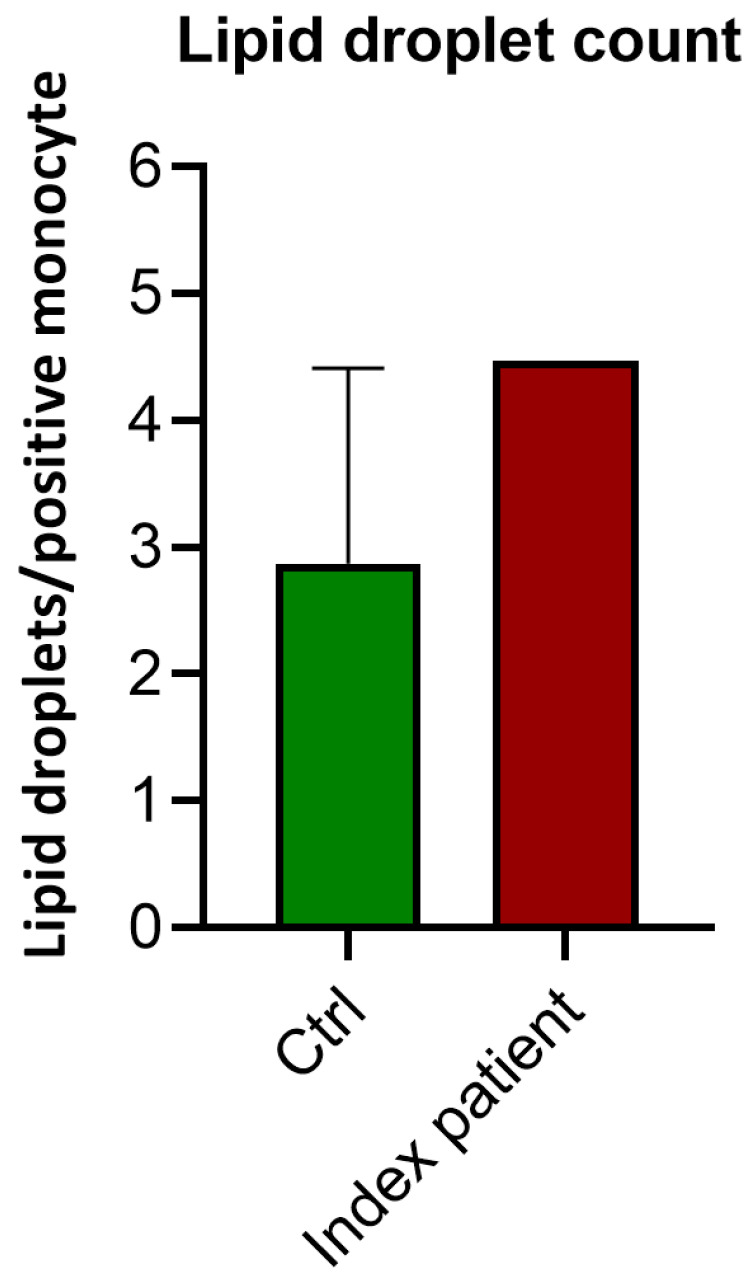 Figure 3
Figure 3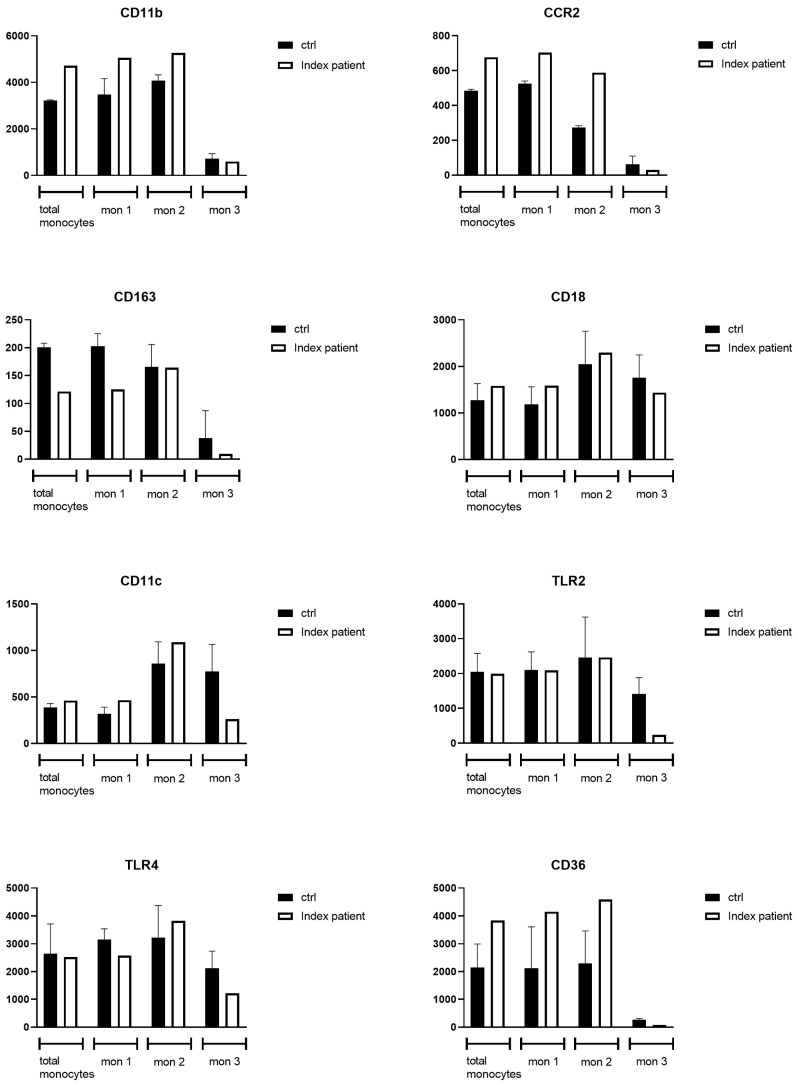 Figure 4
Figure 4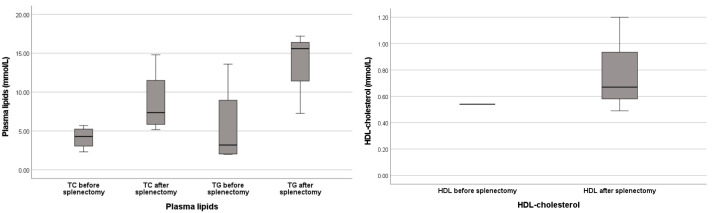 Figure 5
Figure 5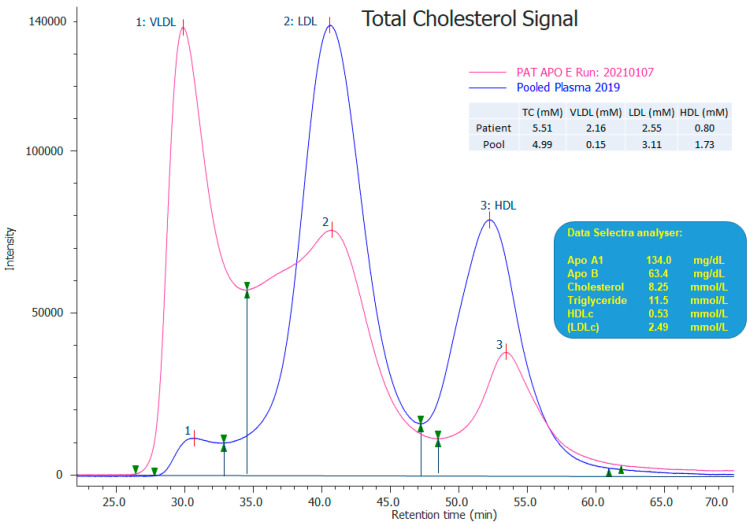 Figure 6
Figure 6| Patient ID (Reference) | Case Type | Age (Years), Sex | BMI > 25 kg/m2 | Cardiovascular Involvement | Splenomegaly | Plasma Lipid | Foam Cells | |||
|---|---|---|---|---|---|---|---|---|---|---|
| TG | TC | |||||||||
|
|
|
|
|
|
|
|
| 11.7 | 7.2 | Y (Bone marrow and spleen) |
|
| Brother of index patient | 26, M | p.(Gly171Asp)/p.(Arg202Cys) (ε1) | p.(Gly171Asp)/p.(Arg202Cys) (ε1) | Y | N | Y | 3.2 | 9.2 | NM |
|
| Proband | 76, M | p.(Leu167del) (ε3) | p.(Arg202Cys) | Y | Severe ischemic heart disease; essential hypertension; systolic ejection murmer (grade 2/6) | Y | 16.2 | 6.2 | N |
|
| Proband | 49, M | p.(Leu167del) (ε3) | p.(Arg202Cys) | Y | N | Y | <2.0 | Normal | Y (Spleen) |
|
| Proband | 47, M | p.(Leu167del) (ε3) | p.(Arg202Cys) | Y | N | Y | 13.6 | 4.8 | Y (Bone marrow and spleen) |
|
| Brother of C | NR, M | p.(Leu167del) (ε3) | WT | NR | Ischemic heart disease | Y | 1.9 | 5.7 | NR |
|
| Proband | 29, M | p.(Leu167del) (ε3) | WT | Y | Ischemic heart disease | Y | 2.1 | 2.3 | Y (Spleen) |
|
| Mother of E | NR, F | p.(Leu167del) (ε3) | p.(Arg202Cys) | NR | Ischemic heart disease | Y | 1.7 | 4.6 | NR |
|
| Proband | 49, M | p.(Leu167del) (ε3) | WT | Y | Ischemic heart disease | Y | 4.3 | 3.8 | Y (Spleen) |
| Patient ID (Reference) | Case Type | Age (Years), Sex | BMI > 25 kg/m2 | Cardiovascular Involvement | Splenomegaly | Plasma Lipid | Foam Cells | |||
|---|---|---|---|---|---|---|---|---|---|---|
| TG | TC | |||||||||
|
|
|
|
|
|
|
|
| 11.7 | 7.2 | Y (Bone marrow and spleen) |
|
| Brother of index patient | 26, M |
|
| Y | N | Y | 3.2 | 9.2 | NM |
|
| Proband | 49, M | WT | p.(Gly171Asp)/p.(Arg202Cys) | Y | Inverted T-waves on ECG | NR | 12.0 | 5.3 | NR |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Proband | 42, F | p.(Gly171Asp)/p.(Arg202Cys) | p.(Arg202Cys) | Y | N | NR | 5.7 | 9.4 | NR |
|
| Son of K | 15, M | p.(Gly171Asp)/p.(Arg202Cys) | p.(Arg202Cys) | Absence significant obesity reported | NR | NR | 4.2 | 7.7 | NR |
|
| Son of K | 13, M | p.(Gly171Asp)/p.(Arg202Cys) | WT | NR | NR | NR | 0.5 | 4.8 | NR |
|
| Son of K | 9, M | p.(Gly171Asp)/p.(Arg202Cys) | p.(Arg202Cys) | NR | NR | NR | 1.6 | 4.5 | NR |
|
| Uncle of K | 78, M | p.(Gly171Asp)/p.(Arg202Cys) | p.(Arg202Cys) | NR | N | NR | 2.9 | 9.6 | NR |
|
|
|
|
|
|
|
|
|
|
|
|
- —Stichting Metakids
- —United for Metabolic Diseases
- —Dutch Heart Foundation
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLysosomal Storage Disorders Research · Pancreatitis Pathology and Treatment · Sphingolipid Metabolism and Signaling
1. Introduction
Splenomegaly has a broad differential diagnosis, including infections, hematologic malignancies, hepatic disease, focal lesions in the spleen and autoimmune disease [1]. However, when the most common causes are ruled out and splenomegaly persists, the suspicion of an inherited metabolic disorder (IMD) grows. The most frequently diagnosed group of IMDs in adult patients presenting with unexplained persistent splenomegaly are the lysosomal storage diseases (LSDs) (see Appendix A Table A1 for a full overview of all IMDs associated with splenomegaly) [2]. LSDs are a heterogeneous group of disorders resulting from variants in genes encoding lysosomal degradative enzymes, associated activator proteins or lysosomal transporters that are essential for exporting degraded products from the lysosome [3]. Lysosomal enzyme malfunction leads to the buildup of specific substrates, eventually resulting in lysosomal dysfunction [2]. The specific type and pattern of affected organ systems points towards the most likely diagnosis (e.g., pulmonary involvement for Niemann–Pick type B (NPB; OMIM #607616) and corneal opacities for LCAT deficiency (OMIM #606967)) [2].
A subset of IMDs that can present with splenomegaly in adulthood are also characterized by marked changes in the lipoprotein profile (e.g., Tangier disease (OMIM #205400) and cholesterol ester storage disease (OMIM #278000)). The combination of unexplained dyslipidemia and splenomegaly is therefore a strong indicator for an underlying genetic cause [1]. A combination of more general biomarkers (e.g., chitotriosidase activity, a macrophage activation marker which is elevated in, among other diseases, Gaucher (OMIM #230800), Niemann–Pick type B and C disease (NPC; OMIM #257220)) and highly specific biomarkers (e.g., glucosylsphingosine for Gaucher disease) can contribute to identifying the correct diagnosis. The diagnosis can subsequently be confirmed by the measurement of the activity of specific enzymes and genetic testing. In the group of “splenomegaly-causing LSD”, Niemann–Pick disease type C takes a special place since it is not a specific enzyme deficiency but a cholesterol trafficking disorder. It shares part of its clinical presentation with other LSDs: splenomegaly, elevated macrophage activation markers and, in some cases, the presence of foamy macrophages in bone marrow. For Niemann–Pick disease type B and C, N-palmitoyl-O-phosphocholine-serine (PPCS, initially named lysosphingomyelin-509) is a sensitive biomarker, especially when grossly elevated. To subsequently discriminate between these two types of Niemann–Pick disease, the lyso-sphingomyelin value can be determined, which is markedly increased in patients with Niemann–Pick disease type B [4]. PPCS is detectable in various matrices (cerebrospinal fluid, blood, and liver products) and belongs to a class of newly characterized lipids. Its biosynthetic pathway, however, has yet to be uncovered [5]. Formerly thought to be NPC specific, this marker has recently also been shown to be elevated in several other disorders, including a congenital disorder of glycosylation, ATP6AP1-CDG (OMIM #300197) [5]. In this article, we show that the differential diagnosis for an adult presenting with a macrophage storage disorder phenotype, including elevated levels of PPCS, should be broadened with dyslipidemic splenomegaly, caused by pathogenic variants in APOE (Figure 1). We base this conclusion on the genetic and biochemical findings in our index patient, a 37-year-old man with splenomegaly, his brother and functional studies performed with the macrophages of the index patient. In addition, we performed a meta-analysis of comparable cases reported in literature to find additional evidence of the relationship between pathogenic APOE variants and the inherited dyslipidemic splenomegaly phenotype.
2. Materials and Methods
2.1. Subjects
2.1.1. Brief Description of the ZOEMBA Study
The ZOEMBA (Dutch full name: ZOektocht naar Erfelijke MetaBole Aandoeningen) is a prospective, diagnostic, multicentre cohort study. Genomics (whole-exome sequencing (WES) reanalysis/whole-genome sequencing (WGS)) and untargeted metabolomic technologies (next-generation metabolomics screening in both plasma and blood spots [6,7] are combined with extensive phenotyping to find the genetic cause in patients with an unexplained phenotype suggestive of an IMD in whom standard of care diagnostics did not yield a diagnosis.
2.1.2. Ethics Approval
The ZOEMBA study protocol and informed consent form was reviewed and approved by the medical ethics committee of the Amsterdam UMC (NL67721.018.19) and registered at https://clinicaltrials.gov (accessed on 16 January 2025) (NCT06200142). The study complied with the Declaration of Helsinki, Good Clinical Practice, and the regulatory requirements in the Netherlands. The authors have obtained written consent forms from both patients (index patient and his brother).
2.1.3. Beacon Protocol
All three control subjects (matched to the index patient for both age and gender) were recruited via the “Beacon protocol”. This collaboration initiative of the biomedical departments in the Amsterdam UMC, reviewed and approved by the Amsterdam UMC medical ethics committee, recruits healthy volunteers for bodily fluid donations. All control subjects signed informed consent via the Beacon protocol prior to inclusion.
2.1.4. Genomic Analysis
WES was performed for the index patient on an Illumina HiSeq4000 sequencer (Illumina, San Diego, CA, USA). Reads were mapped along the GRCh37 (HG19) reference genome using BWA v0.7.12 and duplicate marked using Picard v1.90. Subsequently, variant calling of single-nucleotide variants (SNVs) and small indels was carried out using GATK v.3.4-46. Short-tandem repeats (STRs) were analyzed using Expansion Hunter v3.1.2. with default settings. Variant annotation was performed using a custom diagnostic annotation pipeline. Low-quality variants and variants with a frequency >1% in dbSNP, GnomAD v2.1 or the in-house database were filtered out. The data were first analyzed in a diagnostic setting using a virtual customized gene panel including SMPD2, SMPD3, SMPD4, CTSB and CTSL. Six months later, reanalysis of the WES data was performed as part of the ZOEMBA study, where an open exome analysis was performed. In the reanalysis, all nonsense variants and missense variants with a CADD score > 20, phyloP > 2.7 or spliceAI ≠ 0 were prioritized. Subsequently, for known disease genes, the genotype–phenotype correlation was studied.
2.1.5. Nomenclature of APOE Isoforms
APOE (NM_001302688.2) is known to have two polymorphisms, rs429358 and rs7412, resulting in three common protein isoforms (APOE-ε2, APOE-ε3 or APOE-ε4). The APOE-ε3 (Cys156/Arg202, formerly annotated as Cys112/Arg158) is considered the wild-type form and accounts for approximately ~60–75% of all alleles [1]. The other APOE isoforms have less affinity for lipoprotein receptors on cells, thus are perceived as less functional or even defective [1,8,9]. APOE-ε2 (Cys156/Cys202, formerly annotated as Cys112/Cys158) constitutes ~10% of all alleles, and functional tests have demonstrated that APOE-ε2 binds with <2% affinity to the LDL-receptor compared to APOE-ε3 [10,11,12]. However, despite ~1% of the population being homozygous for APOE-ε2, only ~5% of these homozygotes develop dysbetalipoproteinemia, mostly in the presence of other risk factors for dyslipidemia [13]. APOE-ε4 (Arg156/Arg202, formerly annotated as Arg112/Arg158), accounting for approximately 15% of all alleles, has been associated with an increased risk for Alzheimer’s disease and will not be discussed in this paper [12,14].
On top of these relatively common isoforms, additional rare (pathogenic) variants in APOE can present, with a possible additive negative effect on the functionality of the APOE protein. A rare occurrence is the presence of a second variant in the APOE-ε2 variant, causing a second amino acid change (glycine to asparagine at position 171, formerly annotated as 127) in APOE-ε2 isoform (Cys156/Cys202), thus yielding a protein with two amino acid changes compared to wild-type APOE-ε3, which is referred to as the APOE-ε1 isoform [9,15]. Similarly to APOE-ε2, APOE-ε1 has a reduced binding ability (<4%) to the LDL-receptor compared to APOE-ε3 [9]. The APOE-ε1 allele is also known as the “Weisgraber allele”. Its presence (in the heterozygous state) has been associated with dysbetalipoproteinemia [9,15,16,17].
2.2. Functional Assays in Monocytes of the Index Patient and Three Healthy Control Subjects
2.2.1. Nomenclature of Monocyte Fractions
Human monocytes can be subdivided into three fractions, each having different functions in the immune response, based on their expression of surface markers: classical, intermediate and non-classical monocytes [18]. CD14^++^CD16 monocytes are classified as classical, CD14^++^CD16^+^ are intermediate and CD14^+^CD16^+^ are classed as non-classical. Classical monocytes are involved in phagocytosis, adhesion, migration and anti-microbial responses. Intermediate monocytes regulate apoptosis, transendothelial migration and antigen presentation. Lastly, the function of non-classical monocytes consists of complement and FcR-mediated phagocytosis, trans-endothelial migration, adhesion and anti-viral responses [18].
2.2.2. Intracellular Lipid Droplet Accumulation
Nile Red (9-diethylamino-5H-benzo [α]phenoxazine-5-one) was dissolved in DMSO (318 µg/mL => 1 mM) and filtered through a 0.22 µM syringe to reduce background and create a homogenous solution. The stock solution was diluted in PBS to a final concentration of 10 µM (protect from light). Microscope glass was coated with fibronectin after drawing a circle (diameter 1.5 cm) with a DAKO pen for at least 1 h. Cells were added 100–200 µL (0.5 × 10^5^ cells in total) and incubated for 1 h at 37 °C, 5% CO_2_. Cells were then fixed for 15 min at room temperature (4% formaldehyde). Cells were washed with PBS and stored at 4 °C. Cells were permeabilized for 5 min with 0.1% Triton X-100, once washed with PBS and incubated with 3.3 µM Nile Red for 15 min. Cells were washed with PBS and mounted (DAKO fluorescent mounting media). Monocytes were imaged with the Leica TCS SP8 Confocal microscope (Leica Microsystems GmbH, Mannheim, Germany) (63× Oil objective was used (phospholipids were excited at 590 (600–700 nm) and neutral lipids at 488 (500–580 nm). Quantification was performed by counting the total number of monocytes with lipid droplets per field of view (FOV), as well as the number of lipid droplets per positive monocyte in 6–10 FOVs. To visualize all lipid droplets, z-stack images of 1 µM per stack were made (1024 pixels × 1024 pixels).
2.2.3. Flow Cytometry
Whole blood was collected from the index patient and age- and gender-matched healthy control subjects. Red blood cells were lysed using red blood cell lysis buffer 10× (eBioscience, San Diego, CA, USA) (followed by staining of white blood cells for the surface markers CCR2, CD11c, CD36, CD29, CD18, CX3CR1, TLR2, CD11b, SR-A, TLR4, HLA-DR, CD14, CD16 and IVIG (See Appendix B Table A2|Antigen overview). Fluorescent intensity was measured using a FACS CANTO II (BD) and analyzed with FlowJo software version 10.6. Subsequently, the monocyte area was gated based on forward and side scatter, HLA-DR and CD14^+^ and/or CD16^+^. Then, monocytes were classified as classical (CD14^++^CD16^−^), intermediate (CD14^++^CD16^+^) or non-classical (CD14^+^CD16^+^). The expression of cell surface markers was calculated as the delta geometric mean (∆GM). ∆GM = GM surface staining—GM unstained control.
2.3. Literature Search
A retrospective case-report-based meta-analysis was performed, looking for case descriptions of patients with splenomegaly and variants in APOE. In addition, we searched for reports on individuals with heterozygous or homozygous Weisgraber allele(s). The meta-analysis was performed according to the “Preferred Reporting Items for Systematic Reviews and Meta-Analyses statements” [19,20]. We searched for eligible articles using two combinations of Medical Subject Headings (MeSH) and free text from the inception of these databases to 29 March 2023. We also hand-searched references of all included studies to identify review articles and related meta-analyses. The search was performed with the following terms: ((“Apolipoproteins E”[Mesh]) AND “Splenomegaly”[Mesh]) AND “Dyslipidemias”[Mesh] “APOE” AND “Splenomegaly”[MESH terms] and (((“Apolipoproteins E”[Mesh]) AND “Weisgraber”) AND Apolipoprotein E1). E.A.F. conducted the literature search and E.A.F. and M.L reviewed the titles, abstract, and full-text articles to determine if they met the inclusion criteria (APOE variants AND splenomegaly OR Weisgraber allele/APOE-ε1 WITH/WITHOUT splenomegaly). Ambiguities were resolved through discussion with co-author M.L. Only publications written in English were included (See Figure 2, PRISMA flowchart).
2.4. Statistical Analysis
Boxplots and statistical comparisons were constructed using IBM SPSS statistics, version 26. Statistical comparisons between the non-parametric groups (literature meta-analysis) were performed by a Wilcoxon Signed Rank test. A two-sided p-value of ≤ 0.05 was considered significant.
3. Results
3.1. Case Report
A 37-year-old male (index patient, Table 1 and Table 2) of Moroccan descent and from consanguineous parents presented with acute sensations of pain in the upper left quadrant of the abdomen due to splenic infarction in a grossly enlarged spleen. Prior medical history included hypersplenism since early childhood (at this time no cause was established and monitoring was discontinued in his teens) and obesity (BMI 33 kg/m^2^). The family history was positive for coronary artery disease (CAD). Six out of 14 siblings of the patient’s father suffered from CAD from a young age, including the father himself, who had died prematurely from acute pancreatitis of an unknown etiology (triglyceride levels were not determined at the time of his hospital admission). Common causes of splenomegaly, such as viral infections, were excluded, and a bone marrow biopsy was performed because of suspicion of a hematological malignancy. The histological examination showed macrophage foam cells, suggestive of a LSD. The lysosphingolipid profile in plasma showed a pronounced elevation of N-palmitoyl-O-phosphocholine-serine (PPCS; 547 nmol/L, reference range 1.3–32.2 nmol/L) and a marginally elevated lyso-sphingomyelin (7.4 nmol/L, reference range 0.2–3.0 nmol/L). Furthermore, chitotriosidase activity was elevated (901 nmol/h/mL, reference range 0–90 nmol/h/mL), as were oxysterol levels: 7-ketocholesterol (0.75 umol/L, reference range 0–0.52) and cholestane-3-β,5-alfa,6-β-triol (0.29 µmol/L, reference range 0–0.058). Sphingomyelinase activity was slightly decreased (9 nmol/h/mg, reference range 10–53 nmol/h/mg). Filipin staining in fibroblasts was negative.
The clinical phenotype (enlarged spleen, foamy macrophages in bone marrow) and the metabolite pattern (massively elevated PPCS, elevated chitotriosidase activity and increased oxysterols) resembled that of Niemann–Pick type C; however, no neurological abnormalities (specifically no supranuclear gaze palsy) were found with detailed clinical examination. Additionally, no pathogenic variants were detected in NPC1/NPC2 or SMPD1 by Sanger sequencing and Multiplex Ligation-dependent Probe Amplification (MLPA). Due to the high suspicion of sphingolipid metabolism pathology in the index patient, exome analysis with a virtual customized gene panel was performed (SMPD2, SMPD3, SMPD4, CTSB and CTSL), which did not identify any (potentially) pathogenic variants. Next, an open WES reanalysis was performed as part of the ZOEMBA study. A shortlist of 17 variants was generated with rare (<1% MAF) nonsense variants or missense variants with a CADD score > 20 (see Appendix B Table A3 for a full overview of detected, potentially pathogenic, variants). For known disease genes, the phenotype–genotype correlation was checked, which led to the prioritization of a homozygous missense variant in APOE.
The detected homozygous missense variant c.512G>A p.(Gly171Asp), classified as class 3 (a variant of unknown significance) in ClinVar, was prioritized due to the previously reported association with splenomegaly and foam cells in bone marrow in patients with other APOE variants in literature [8,21]. In addition, the index patient was shown to be homozygous for APOE-ε2 rs7412 c.604C>T p.(Arg202Cys). This second variant was initially not prioritized in the analysis because of the relative high frequency in the healthy population (GnomAD v.2.1.1, MAF 12% heterozygous and 0.5% homozygous); however, since this patient was now homozygous for both variants, and therefore homozygous for the Weisgraber allele (APOE-ε1), all APOE variants were considered interesting for the phenotype (see discussion). Additionally, a variant in NPC1 (NM_000271.5) c.463+19A>G was identified, but this variant has been reported as benign in ClinVar and thus deemed not causative of the phenotype. Due to the identification of the homozygous Weisgraber alleles, a lipid panel was acquired according to the method described by Heidemann et al. [22]; this can only be performed by specialized laboratories using ultracentrifugation. The lipid panel showed (after removal of chylomicrons and compared to pooled plasma of control subjects) a total cholesterol (TC) of 5.5 mmol/L (pool 4.99); very-low-density lipoprotein (VLDL) 2.16 mmol/L (pool 0.15); low-density lipoprotein (LDL) 2.55 mmol/L (pool 3.11); high-density lipoprotein (HDL) 0.80 mmol/L (pool 1.73) (SeeAppendix B Figure A1|Plasma lipid profile). Next, we offered to include both brothers of the index patient in the ZOEMBA study as they exhibited similar phenotypes, to which the youngest brother agreed; his details are discussed below.
The brother (Table 1 and Table 2), a 26-year-old male, had no relevant medical history except for obesity (BMI 30 kg/m^2^). An ultrasound of the spleen revealed a slightly enlarged spleen (12.4 cm, upper limit of normal). Laboratory studies showed significant elevations in total cholesterol (9.0 mmol/L, reference range < 3.4 mmol/L); total triglycerides (3.2 mmol/L, reference range < 2.0 mmol/L); a normal HDL cholesterol level (1.2 mmol/L, reference range > 0.9 mmol/L); elevated PPCS (81.3 nmol/L, reference range 1.3–32.2 nmol/L) and lyso-sphingomyelin (4.9 nmol/L, reference range 0.2–3.0 nmol/L). Targeted Sanger sequencing of the two homozygous APOE variants and the heterozygous NPC1 variant detected in the index patient were also identified in his brother.
After diagnosis, lipid-lowering medication (Atorvastatin and Ciprofibrate) was started in both brothers. Additionally, an energy-restricted and saturated-fat-restricted diet and lifestyle changes (i.e., regular exercise and cessation of smoking) were advised. The patients were contra-indicated for splenectomy. Additionally, the whole family was offered genetic counseling.
3.2. Functional Studies in Monocytes of Index Patient
3.2.1. Lipid Droplet Count
In order to confirm that the disruption in APOE function leads to lipid accumulation in monocytes, we compared the monocytes of the index patient to those of age- and gender-matched control subjects. The lipid droplet per monocyte count was higher in the index patient (4.47 lipid droplets/monocyte) compared to the three healthy control subjects (median of 2.87 lipid droplets/monocyte (range 2.29–4.41) (Figure 3).
3.2.2. Flow Cytometry Analysis of Monocytes
To study the effect of the lipid accumulation in the monocytes of the index patient, the activation state was analyzed by measuring the expression of monocyte surface markers. Flow cytometry (FACS) results (Figure 4) (see Appendix C for a full overview of all FACS data) showed an increased expression of CD11b, CCR2 and CD36 on the monocytes of the index patient compared to those of healthy control subjects (n = 3), especially in the classical and intermediate monocyte fraction. The CD11b, CCR2 and CD36 expression levels were 5059 ∆GM, 703 ∆GM, 4151 ∆GM in the index patient on the mon 1/classical monocyte fraction, compared to 3620 ∆GM (range 2746–4086), 527 ∆GM (range 511–540) and 2118 ∆GM (range 1655–3610) for the healthy control subjects. CD11b, CCR2 and CD36 showed an expression of 5262 ∆GM, 588 ∆GM, 4594 ∆GM in the index patient on mon 2/intermediate monocyte fraction, compared to an expression of 4006 ∆GM (range 3845–4345), 274 ∆GM (range 204–283) and 1636 ∆GM (range 1612–3644) for the healthy control subjects, respectively. The expression of the anti-inflammatory marker CD163, was reduced in patient monocytes, especially in classical (125 ∆GM) and non-classical (9 ∆GM) monocyte fractions compared to the expression in the classical (207 ∆GM; range 178–223) and mon 3/non-classical (23 ∆GM; range −2.0–93) monocyte fractions of the healthy control subjects [23]. The expression of CD11c and the expression of the pattern recognition receptors, TLR2 and TLR4, were reduced in the patient’s non-classical monocyte fractions only, 262 ∆GM, 241 ∆GM, 1213 ∆GM, respectively, compared to the median expression in the non-classical monocytes of the healthy control subjects of 706 (range 523–1094), 1152 (range 1140–1954), 2427 (range 1399–2519) ∆GM, respectively.
3.3. Meta-Analysis of Literature
To find additional evidence for the relationship between pathogenic APOE variants and the inherited dyslipidemic splenomegaly phenotype present in our index patient, a meta-analysis of cases reported in the literature was conducted. As shown in Figure 2, a total of seven articles were identified in the initial search. One article was excluded after title and abstract screening because it reported on a different gene. After a full-text assessment of the remaining six articles, all fulfilled the inclusion criteria and were included in this meta-analysis. Two more articles were included by hand-searching the references of the initial set of included articles. The eight analyzed articles reported on a total of seven patients with splenomegaly caused by the APOE variant (s). Nine of the described patients presented with APOE-ε1; three patients were homozygous and six were heterozygous.
A total of six patients presented APOE variants in combination with splenomegaly: two patients had homozygous APOE-ε1 variants, and all seven patients who were previously reported on in literature showed the same deletion of leucine 167 (NM_000041) on APOE-ε3 (Table 1). Four out of seven of these patients (individuals A–C and F) also carried an APOE-ε2 isoform, which, in the adult males, generally led to higher levels in triglycerides and/or total cholesterol, even after the initiation of cholesterol-reducing treatment (with the exception of patient B). Seven out of nine patients suffered from either overweight or obesity (BMI ≥ 25 kg/m^2^) (both of our patients and individuals A–C, E and G). Information on BMI was not reported for the remaining two patients. Eight out of nine individuals (our two patients and individuals A, C–G) with splenomegaly showed dysbetalipoproteinemia (defined according to Berberich et al. [24] as LDL-C > 3.4 mmol/L and/or TG > 2.0 mmol/L and/or HDL-C < 0.9); these were mainly adult males. Five out of nine patients (individuals A and D–G), suffered (severe) ischemic heart disease from a relatively young age. Four out of the nine patients (individuals B, C, E and G) underwent splenectomy, and all of these individuals showed infiltration of sea blue histiocytes (foam cells) in spleen tissue. One of these patients also showed infiltration of foam cells in bone marrow. After splenectomy, median triglyceride values rose from 3.2 mmol/L to 16.4 mmol/L. The lipid profiles of individuals B, C, E and G, before and after splenectomy, are summarized in Figure 5.
Eleven patients carried one or two APOE-ε1 alleles (our two patients and individuals H-P; details in Table 2); in none of the patients reported in literature, spleen sizes were assessed. Five out of eleven individuals were homozygous for the APOE-ε1 allele (our two patients and individuals I, J and P). The following characteristics seem to be associated with aggravated dyslipidemia: adulthood, male gender and homozygosity (as opposed to heterozygosity) for the APOE-ε1 allele. Obesity also seems a predisposing factor for developing more severe dyslipidemia (with or without splenomegaly). This is in line with a higher risk of dyslipidemia in APOE-ε2 homozygous individuals in the presence of obesity [25,26].
4. Discussion
In this article, we discuss a patient presenting with splenomegaly and several features indicative of a lysosomal storage disorder. The patient was eventually diagnosed with a macrophage storage disorder resulting from lipid accumulation due to a combination of a relatively common homozygous APOE isoform (APOE-ε2), combined with homozygosity for a rare amino acid change p.(Gly171Asp). This alteration of the APOE-ε2 isoform results in an isoform known as APOE-ε1, also referred to as the Weisgraber allele. Thus, the reported index patient is homozygous for APOE-ε1. The clinical consequences of heterozygosity for APOE-ε1 have been reported in the literature (Table 2), but only three individuals homozygous for APOE-ε1 were identified by our literature search, with limited details on the phenotypes of these individuals being found (Table 2, cases I, J and P). We suggest naming the lipid storage disorder caused by disruptive APOE variants ‘inherited dyslipidemic splenomegaly’, a slight variation on the name of the lipidemic splenomegaly used by Okorodudu et al. in 2013 [1].
To confirm the causal relationship between these changes in APOE and the established macrophage storage phenotype (enlarged spleen, foamy macrophages in bone marrow, elevated chitotriosidase activity), as well as to decipher the pathomechanism, we conducted several functional studies in monocytes. These showed lipid accumulation in monocytes of the APOE-ε1 homozygous index patient. The patient’s monocytes showed an increased expression of CD11b, CCR2 and CD36, predominantly on classical monocytes. This increased expression could functionally lead to increased monocyte adhesion and migration into the intima. As a result of progressive lipid accumulation and activation, these macrophages convert into foam cells and can accumulate in tissues such as the spleen [27,28,29]. Of specific interest is the upregulation of CD36 on the surface of the patient’s classical and mon 2/intermediate monocyte fraction, given the role of CD36 in the uptake of oxidized LDL particles and entrapment of macrophages within atherosclerotic plaques, resulting in foam cell formation [30]. In addition, the expression of the anti-inflammatory marker CD163 in the patient’s classical and mon 3/non-classical monocyte subfractions was reduced. CD163 deficiency has been directly correlated to increased foam cell formation and plaque progression [31]. Moreover, expression of CD163 negatively regulates the release of interleukin-10 (IL-10), which in mouse models has been associated with reduced atherogenesis and an improvement of the stability of atherosclerotic plaques [32]. Conversely, the expression of TLR2 and TLR4 on monocytes (both increased during the development of atherosclerosis) was reduced in the patient’s non-classical monocyte fractions; however, this has been reported before in patients with chronic inflammation [33].
The findings from our literature study further support a causal connection between pathogenic homozygous and heterozygous rare variants in APOE, with a significant negative impact on APOE function and the presenting phenotype (dysbetalipoproteinemia and splenomegaly) (Table 1). In addition, this macrophage activation profile is consistent with an increased risk of early atherosclerosis, which is compatible with the finding of early-onset ischemic heart disease in the patients with p.(Leu167del) (APOE-ε3) reported in literature. Both the findings in our two patients and their family, as well as those reported in literature, suggest that obesity is an important modifier of the severity of the phenotype in individuals who are either heterozygous or homozygous for the APOE-ε1 variant [25,26]. Obesity is known to alter lipid homeostasis, leading to an increased amount of triglyceride-rich lipoprotein particles in plasma, which are known to be cleared less efficiently in patients with disruptive APOE variants [34,35]. The additional predisposing factors for severe dyslipidemia in these patients appear to be the type of genetic variant (i.e., heterozygous vs. homozygous variants and location of the variant/residual APOE function), male gender and a post-splenectomy status [22,36,37]. The impact of these factors, as well as the influence of other genetic modifiers, will need to be confirmed in future studies with larger patient cohorts.
Diagnosing inherited dyslipidemic splenomegaly is important since it has consequences for the medical management of the index patient, namely treatment of the dyslipidemia, preventing/treating obesity and preventing splenectomy. Establishing a diagnosis also prevents further invasive diagnostics into potential hematological malignancies. Data from literature suggest that weight loss may also lead to reduction in spleen size (individual A, Table 1). Genetic counseling and the identification of other family members at risk is essential, as proper measures can prevent the early onset of atherosclerosis and cardiovascular events related to the dysbetalipoproteinemia caused by both the heterozygosity and homozygosity of APOE-ε1, as demonstrated by the index patient’s family history.
It is important to note that splenectomy is contra-indicated in this patient group. In all reported cases with APOE variants in which splenectomy was performed, the dyslipidemia was aggravated after the procedure (Figure 5 and Ai et al. [38]). This is in line with findings from other storage disorders, e.g., Gaucher disease and Niemann–Pick type B disease, where the manifestations in other organs (e.g., bone marrow or lungs) worsen after removal of the spleen, suggesting that the spleen functions as a sponge for the storage of excessive macrophages and foam cells [39,40,41]. The combined information from the index patient’s plasma profile (Appendix B Figure A1) and functional tests support the idea that there is a defect in both the clearance of VLDL from plasma whilst, at the same time, macrophages are more prone to ingest the defective APOE particles, together aggravating the dyslipidemic phenotype (Figure 1).
Although malfunctioning APOE isoforms are currently primarily perceived as risk factors for cardiovascular disease, APOE knock-out mice have confirmed a direct correlation between the loss of APOE and the development of splenomegaly [42]. Furthermore, it has been shown that some (both heterozygous and homozygous) APOE variant combinations are disruptive enough to lead to the development of a lipid storage phenotype in humans, characterized by dysbetalipoproteinemia, and splenomegaly, caused by accumulation of foam cells/sea-blue histiocytes in the spleen [1,8,12,21,43]. In the current report, we show that homozygosity of APOE-ε1 can present with splenomegaly, where the human phenotype shows considerable overlap with lysosomal storage disorders, including the elevation of biomarkers (e.g., chitotriosidase activity, elevated in Gaucher and Niemann–Pick disease type B, and oxysterols, elevated in Niemann–Pick disease type B and C). The marked elevation of plasma PPCS in particular is important, as this was previously thought to be a Niemann–Pick type B and C specific marker but has now been shown to be elevated in several disorders that lead to the storage of lipids in macrophages [4,5,44,45].
5. Conclusions
In summary, patients that are under evaluation for a lysosomal storage disorder may actually suffer from dyslipidemic splenomegaly due to disruptive, heterozygous or homozygous APOE variants. This condition should therefore be added to the differential diagnosis, with a plasma lipid profile consistent with a dysbetalipoproteinemia serving as a diagnostic biomarker.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Okorodudu D.E. Crowley M.J. Sebastian S. Rowell J.V. Guyton J.R. Inherited lipemic splenomegaly and the spectrum of apolipoprotein E p.Leu 167del mutation phenotypic variation J. Clin. Lipidol.2013756657210.1016/j.jacl.2013.09.00324314356 · doi ↗ · pubmed ↗
- 2Rajkumar V. Dumpa V. Lysosomal Storage Disease Stat Pearls Stat Pearls Publishing Treasure Island, FL, USA 202333085417 · pubmed ↗
- 3Stirnemann J. Belmatoug N. Camou F. Serratrice C. Froissart R. Caillaud C. Levade T. Astudillo L. Serratrice J. Brassier A. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments Int. J. Mol. Sci.20171844110.3390/ijms 1802044128218669 PMC 5343975 · doi ↗ · pubmed ↗
- 4Voorink-Moret M. Goorden S.M.I. van Kuilenburg A.B.P. Wijburg F.A. Ghauharali-van der Vlugt J.M.M. Beers-Stet F.S. Zoetekouw A. Kulik W. Hollak C.E.M. Vaz F.M. Rapid screening for lipid storage disorders using biochemical markers. Expert center data and review of the literature Mol. Genet. Metab.2018123768410.1016/j.ymgme.2017.12.43129290526 · doi ↗ · pubmed ↗
- 5Dang Do A.N. Chang I.J. Jiang X. Wolfe L.A. Ng B.G. Lam C. Schnur R.E. Allis K. Hansikova H. Ondruskova N. Elevated oxysterol and N-palmitoyl-O-phosphocholineserine levels in congenital disorders of glycosylation J. Inherit. Metab. Dis.20234632633410.1002/jimd.1259536719165 PMC 10023375 · doi ↗ · pubmed ↗
- 6Coene K.L.M. Kluijtmans L.A.J. van der Heeft E. Engelke U.F.H. de Boer S. Hoegen B. Kwast H.J.T. van de Vorst M. Huigen M. Keularts I. Next-generation metabolic screening: Targeted and untargeted metabolomics for the diagnosis of inborn errors of metabolism in individual patients J. Inherit. Metab. Dis.20184133735310.1007/s 10545-017-0131-629453510 PMC 5959972 · doi ↗ · pubmed ↗
- 7Haijes H.A. Willemsen M. Van der Ham M. Gerrits J. Pras-Raves M.L. Prinsen H. Van Hasselt P.M. De Sain-van der Velden M.G.M. Verhoeven-Duif N.M. Jans J.J.M. Direct Infusion Based Metabolomics Identifies Metabolic Disease in Patients’ Dried Blood Spots and Plasma Metabolites 201991210.3390/metabo 901001230641898 PMC 6359237 · doi ↗ · pubmed ↗
- 8Nguyen T.T. Kruckeberg K.E. O’Brien J.F. Ji Z.S. Karnes P.S. Crotty T.B. Hay I.D. Mahley R.W. O’Brien T. Familial splenomegaly: Macrophage hypercatabolism of lipoproteins associated with apolipoprotein E mutation [apolipoprotein E (Δ149 Leu)]J. Clin. Endocrinol. Metab.2000854354435810.1210/jcem.85.11.698111095479 · doi ↗ · pubmed ↗