CCR2 Regulates Referred Somatic Hyperalgesia by Mediating T-Type Ca2+ Channel Currents of Small-Diameter DRG Neurons in Gastric Ulcer Mice
Ziyan Yuan, Huanhuan Liu, Zhijun Diao, Wei Yuan, Yuwei Wu, Simeng Xue, Xinyan Gao, Haifa Qiao

TL;DR
This study shows that CCR2 regulates referred pain by altering calcium channels in nerve cells of mice with stomach ulcers.
Contribution
CCR2's role in modulating T-type Ca2+ channels to cause referred somatic hyperalgesia is newly identified.
Findings
Gastric ulcers increase T-type Ca2+ channel and CCR2 co-expression in DRG neurons.
Blocking CCR2 reduces neurogenic inflammation and referred pain in ulcer mice.
CCR2 agonists mimic ulcer-induced changes in calcium channel behavior in normal neurons.
Abstract
Background: Referred pain frequently co-exists with visceral pain. However, the exact mechanism governing referred somatic hyperalgesia remains elusive. Methods: By injecting 20% acetic acid into the stomach, we established a mouse model of gastric ulcer (GU). Hematoxylin and eosin (H&E) staining was used as the evaluation criterion for the gastric ulcer model. Evan’s blue (EB) and von Frey tests detected the somatic sensitized area. The DRG neurons distributed among the spinal segments of the sensitized area were prepared for biochemical and electrophysiological experiments. The CCR2 antagonist was intraperitoneally (i.p.) injected into GU mice to test the effect of blocking CCR2 on somatic neurogenic inflammation. Results: GU not only instigated neurogenic plasma extravasation and referred somatic allodynia in the upper back regions spanning the T9 to T11 segments but also augmented…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
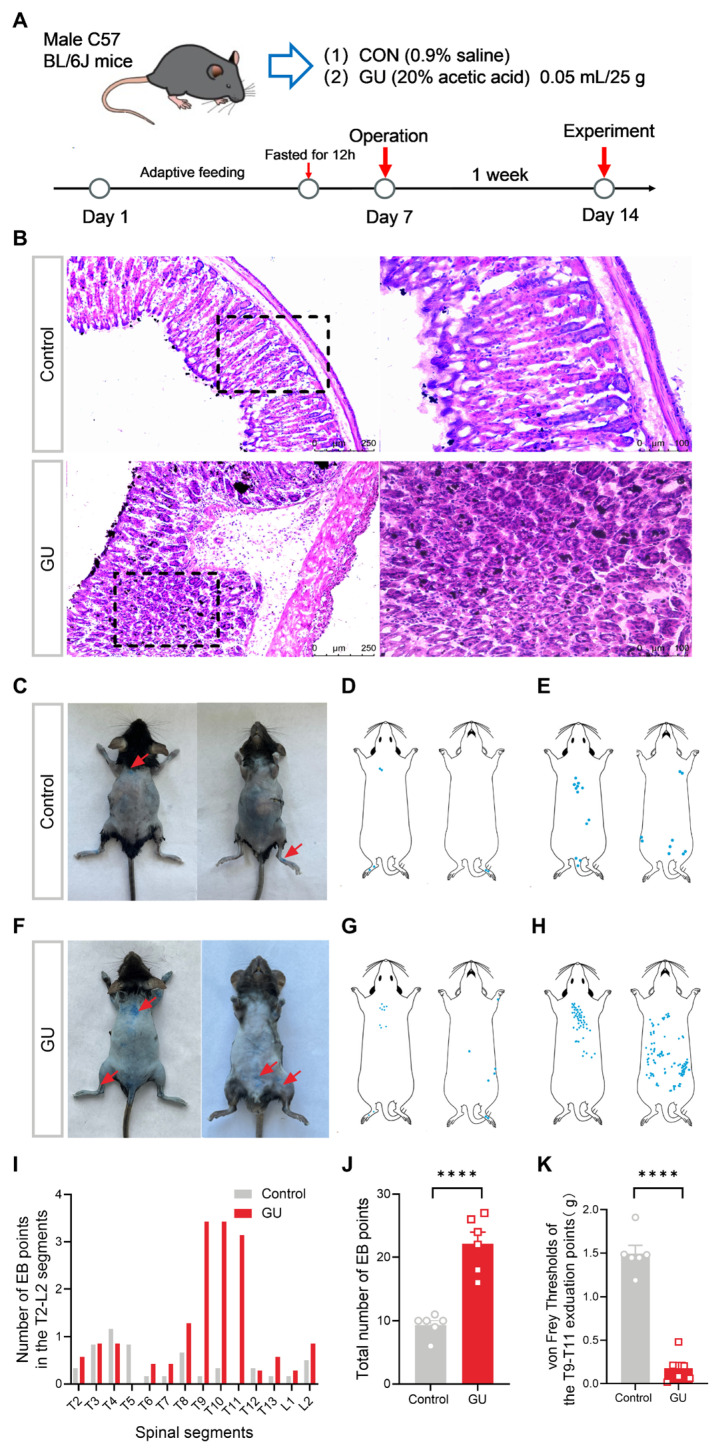 Figure 1
Figure 1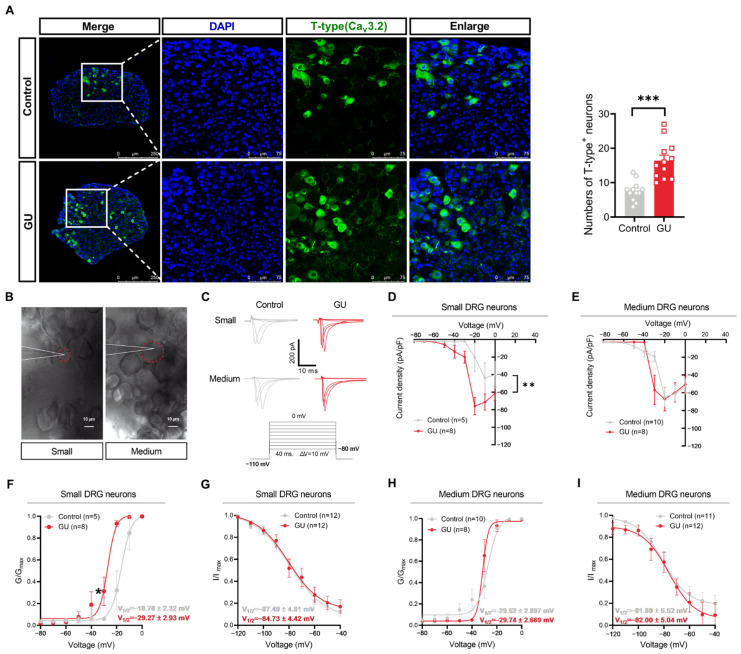 Figure 2
Figure 2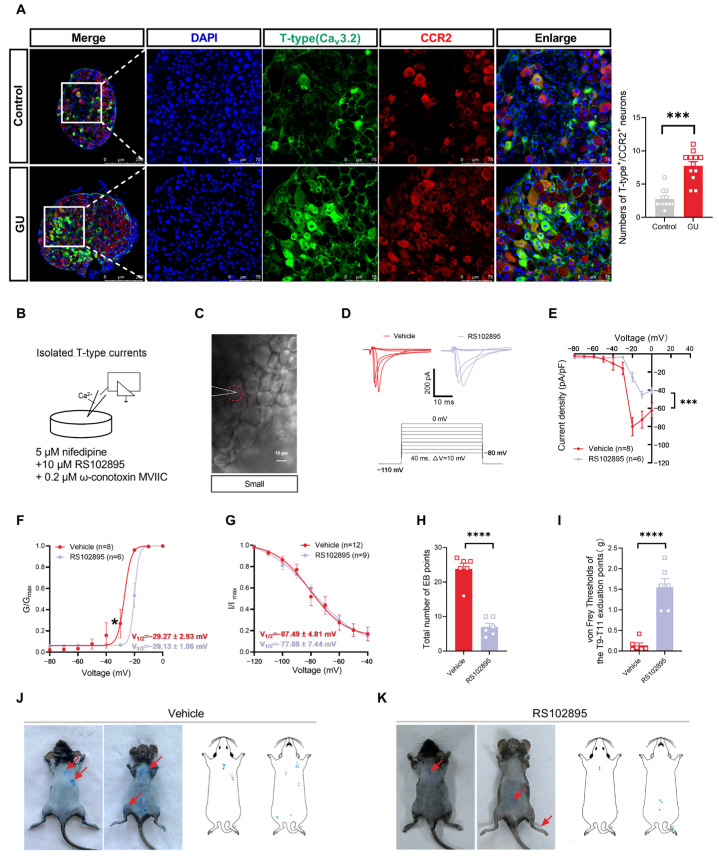 Figure 3
Figure 3- —National Natural Science Foundation of China
- —Natural Key R&D Program of China Grant
- —Key Laboratory Program of Administration of Traditional Chinese Medicine of Shaanxi Province
- —Natural Science Basic Research Program of Shaanxi Province
- —Key Scientific Research Program of Shaanxi Provincial Education Department (Key Laboratory Project)
- —Sci-Tech Innovation Talent System Construction Program of Shaanxi University of Chinese Medicine
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPain Mechanisms and Treatments · Ion Channels and Receptors · Neuropeptides and Animal Physiology
1. Introduction
Visceral pain is uniquely characteristic because it is perceived in areas of the body that are distant from the affected organs, which is termed referred pain. Referred pain frequently co-exists with visceral pain, as seen in examples such as abdomen pain due to irritable bowel syndrome, pelvis pain due to reproductive problems, or the forelimbs and upper back referred pain caused by coronary heart disease [1,2]. This viscerosensory reflex pathway commences with the visceral primary sensory afferent fiber conducting sensory impulses from the viscera to the central nervous system (CNS), ultimately generating somatically referred pain and peripheral sensitization through the mechanisms of the dorsal root reflex (DRR) [3] and axon reflex (AR) [4]. However, the peripheral neurophysiological mechanisms underlying the intricacies of visceral referred pain and sensitization remain largely unclear.
The cell bodies of visceral afferents reside in the dorsal root ganglion (DRG), characterized as pseudo-unipolar neurons, possessing both central and peripheral axonal processes [2]. This distinctive structure allows both the soma and axon to exhibit electrical excitability, thereby facilitating the continuous transmission of sensory information [5]. Therefore, the DRG is essential in transmitting visceral pain signals to the CNS and peripheral nervous system (PNS). Many studies have shown that DRG neurons are implicated in the development of chronic pain, marked by increased excitability and spontaneous ectopic firing [5,6,7,8]. Nevertheless, the ion channel mechanisms and molecular pathways underlying nociceptor hyperexcitability associated with pain remain incompletely elucidated.
Voltage-dependent Ca^2+^ channels (VDCCs) are pivotal to modulating the excitability of sensory neurons under both normal and pathological conditions [9]. These channels can be categorized into high-voltage-activated (HVA, N, P/Q, L, R-types) and low-voltage-activated (LVA, T-type) subtypes [10]. T-type channels distinctively modulate neuronal excitability through their low-voltage activation and window current properties [11], with functional alterations directly impacting pain thresholds. The activation of the T-type channels enhances the excitability of DRG neurons, contributing to mechanical and thermal hypersensitivity [12]. Conversely, the selective in vivo blockade of T-type channels mitigates thermal hyperalgesia and allodynia in rats [13]. Nevertheless, how T-type channels are affected by visceral pain disorders, leading to peripheral sensitization, remains inadequately understood.
T-type Ca^2+^ channels are regulated by different second messenger pathways that respond to the activation of various classes of G protein-coupled receptors (GPCRs) [14]. Several studies suggest that the upregulation of the C-C chemokine receptor 2 (CCR2) expression enhances the excitability of DRG neurons in visceral pain models by sensitizing primary afferent neurons [15]. Additionally, the C-C motif chemokine ligand 2 (CCL2) interacts directly with its receptor CCR2, resulting in increased neuronal firing, which ultimately amplifies nociceptive responses [16,17,18], whereas the CCR2 receptor antagonists partially inhibit T-type channels and elicit analgesia effects [19]. However, it is still not well understood whether the expression of CCR2 on DRG neurons contributes to peripheral sensitization and somatic referred pain hypersensitivity in visceral pain disorders by modulating T-type channels.
In this study, to address these questions, we first established a visceral pain mouse model of gastric ulcers (GU) [20] and identified the referred somatic regions in these mice. Then, behavioral tests were employed to assess somatic hypersensitivity in referred areas of GU mice. Additionally, whole-cell patch-clamping recordings were applied to evaluate the changes in gating properties of T-type Ca^2+^ channels in DRG neurons distributed among the spinal segments of the sensitized regions. With this framework, we investigated the relationship between the role of T-type Ca^2+^ channels and the expression of CCR2 on DRG neurons in GU mice. Finally, we utilized pharmacological methods to evaluate the sufficiency and necessity of T-type Ca^2+^ channels and CCR2 in somatic sensitization and referred pain in GU mice.
2. Materials and Methods
2.1. Gastric Ulcer Mouse Model
Male C57 BL/6J mice, weighing between 20 and 25 g, were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (license number: SCXK-2021-0006, Beijing, China). All animal procedures received approval from the Animal Care Committee of the Shaanxi University of Chinese Medicine (Approval ID: SUCMDL20200420003) and followed the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. The mice were allocated into control (CON) and gastric ulcer (GU) groups for the experiments, as shown in Figure 1A. Ulcers were induced through the administration of acetic acid, following methodologies outlined in previous studies. Under anesthesia, a midline epigastric laparotomy was performed to expose the stomach, into which 50 µL of 20% acetic acid (A6283, Sigma, St. Louis, MO, USA) was injected with a syringe into the sub-serosal layer of the glandular (0.5 mL) [19]. The control group received an equivalent volume of normal saline. Ulcer induction/sham operations were performed by separate investigators. Other experiments were conducted with rigorous randomization and blinded approaches, whereby the experimenter did not know the specific experimental group throughout the procedure.
2.2. Histopathological Examination
One week post operation, the mice were anesthetized and perfused with 0.9% saline injection. Then, the gastric tissues were harvested and immersed in 4% paraformaldehyde (PFA) overnight. The gastric tissues underwent dehydration in a gradient concentration sucrose solution and embedded in OCT. Hematoxylin and eosin (H&E) staining was carried out on frozen sections (thickness 14 μm) using an H&E staining Kit (TOP 0446, Biotopped, Beijing, China). The stained sections were examined under a DM2500 digital light microscope (Leica, Wetzlar, Germany) to assess morphology and signs of inflammation.
2.3. Detection of Sensitized Points
One week post operation, the mice were subjected to deep anesthetization using 2% isoflurane (R510-22-10, RWD, Shenzhen, China) and were depilated. An insulin needle was employed to inject Evan’s blue (EB) dye (50 mg/kg, 100 μL) into the tail vein. Two hours later, the EB exudation was counted, photographed, and recorded on a diagram illustrating the mouse’s somite locations. The number of EB points in the T2-L2 segment as well as the total number of EB points were, respectively, counted in the control and GU groups.
2.4. Behavioral Tests
To determine the referred pain occurred in GU mice, we applied a series of von Frey filaments (ranging from 0.04 to 2.0 g) according to the up–down method previously described [21] to record the mechanical pain threshold on the concentrated blue exudation points of the upper back in mice. Each filament was tested five times for durations of 1 to 2 s. If the mice exhibited a significant escape response, the next lower force filament was applied until two filaments were tested without a positive response. The pressure value of the lowest filament was recorded as the threshold for mechanical pain.
2.5. Preparation of Intact Whole-Mount DRG
The mice were intraperitoneally (i.p.) injected with 25% urethane (51-79-6, Sigma, USA) for anesthetization. The intact whole-mount T9–T11 DRG was isolated from mice according to previously established methodologies [6,22]. In brief, the DRG were bilaterally excised and subjected to enzymatic digestion utilizing a mixture of collagenase (2 mg/mL, 9002-07-7, Sigma, USA) and trypsin (1 mg/mL, 10103586001, Sigma, USA) at 37 °C for 15 min, then incubated in oxygenated artificial cerebrospinal fluid (ACSF) for at least 1 h before recording. The recording chamber was saturated with oxygenated ACSF containing (in mM) the following: 140 NaCl, 5 KCl, 10 Glucose, 10 HEPES, 1 MgCl_2_, and 2 CaCl_2_; in addition, pH was adjusted to 7.4 with NaOH.
2.6. Electrophysiological Recordings
Whole-cell patch-clamp recordings were conducted at ambient temperature using a Digidata 1550B acquisition system and Axopatch 700B amplifier (Molecular Devices, San Jose, CA, USA) with pClamp 10.7 software. The whole-mount DRGs were visually identified with an upright microscope (BX51WI, Olympus, Tokyo, Japan) equipped with infrared differential interference contrast optics (IR-DIC). Series resistance was compensated by at least 75%, and current traces were corrected using online P/4 trace subtraction. The pipette solution contained (in mM): 130 CsCl, 2.5 MgCl_2_, 10 HEPES, 5 EGTA, 3 Na_2_-ATP, and 0.5 Na_2_-GTP; in addition, pH was adjusted to 7.4 with CsOH. To record Ca^2+^ currents, 1 μM of TTX, 5 mM of TEA-Cl, and 2 mM of 4-AP were added to the ACSF to inhibit sodium and potassium currents.
We applied 0.2 μM of ω-conotoxin MVIIC (N- and P/Q-type channel blocker) and 5 μM of nifedipine (L-type channel blocker) to the ACSF, and then used a 40 ms depolarizing step pulse to record currents from −110 to −40 mV in order to isolate T-type Ca^2+^ currents [12]. Activation curves were evoked with voltage steps from −110 mV to test potentials between −80 and 0 mV in 10 mV increments. Inactivation curves were recorded using a 40 ms test pulse to −40 mV after the 1 s conditioning pulses ranging from −120 to −30 mV with 10 mV increments. Current amplitudes were measured from the peak to the end of the depolarizing pulse. Boltzmann distributions characterized the voltage dependence of activation and steady-state inactivation:
Activation: G/G_max_ = 1/{1 + exp [(V_1/2_ − V_m_)/k]}^−1^
Inactivation: I/I_max_ = 1/{1 + exp [(V_m_ −V_1/2_)/k]}^−1^
In these formulas, G_max_ denotes the maximal conductance, I_max_ indicates the maximal current amplitude, V_1/2_ is the voltage for half-activated or inactivated, and k signifies the slope of the distribution.
2.7. Immunofluorescence Labeling
One week post modeling, the mice were anesthetized and underwent cardiac perfusion with saline followed by 4% PFA. The DRG tissues from the T9–T11 segments of the spinal cord were harvested and post-fixed in 4% PFA at 4 °C overnight before being dehydrated in a 30% sucrose solution. Sixteen-micron DRG slices were blocked in phosphate-buffered saline (PBS) with 5% normal donkey serum and 0.5% Triton X-100 for 2 h at room temperature. The sections were then treated with primary antibodies, including mouse anti-Ca_v_3.2 (1:100, NBP1-22444, Novus, Centennial, CO, USA) and rabbit anti-CCR2 (1:100, ab216863, Abcam, Cambridge, UK) incubated at 4 °C for 24 h. Next, three PBS washes were performed on the DRG sections, followed by incubating in secondary antibodies, which include Alexa Fluor 488 (donkey anti-mouse IgG, 1:250, 34106ES60, Yeasen, Gaithersburg, MD, USA) and Alexa Fluor 594 (donkey anti-rabbit IgG, 1:100, 34212ES60, Yeasen). The sections were placed in the darkroom for 2h, after which they were cleaned three more times with PBS and mounted with an anti-fluorescent attenuating sealer containing DAPI. All images were captured with a TCS SP8 confocal microscope (Leica, Germany) and further analyzed using Image J software (Fiji version, NIH). Three sections were randomly selected from each mouse for imaging and quantification.
2.8. CCR2 Antagonist and Agonist Treatment
To test the effect of CCR2 on somatic neurogenic inflammation and referred-mechanical hypersensitivity, mice were administered with an i.p. injection of 5 mg/kg CCR2 antagonist RS102895 (HY-18611, MedChemExpress, Monmouth Junction, NJ, USA) or the same volume of vehicle (10% DMSO) solution one week after operation. The detection of sensitized points and behavioral tests were performed 30 min after drug administration. Additionally, to substantiate the role of CCR2 in DRG during visceral pain, DRG samples were bathed with 100 nM of CCR2 agonist CCL2 (HY-P7764, MedChemExpress, Monmouth Junction, NJ, USA) or 10 μM RS102895 for T-type channel currents recording. All drugs were administered at the same doses as previously reported [23,24].
2.9. Statistical Analysis
Statistical comparisons of data obtained from the detection of sensitized points, behavioral tests, and immunofluorescence labeling in control and GU mice were conducted using the unpaired t-test. A two-way analysis of variance (ANOVA) with the post hoc test was employed to discern variations in current density and the Boltzmann fit curves between different groups. Differences in parameters of the Boltzmann fit formula among groups were evaluated using an unpaired t-test. Data are presented with significance set at p < 0.05.
3. Results
3.1. Neurogenic Inflammation and Referred Mechanical Hypersensitivity in Somatic Regions Induced by Gastric Ulcer
The H&E staining of gastric tissues in the control group displayed normal epithelial cells and no noticeable congestion or edema in the gastric mucosa (Figure 1B). In contrast, exposure to acetic acid resulted in significant gastric tissue damage, manifesting as the loss of epithelial cells, submucosal inflammatory cell infiltration, and reduced mucosal glands (Figure 1B). These results indicate that the injection of 20% acetic acid, but not of saline, is sufficient to induce gastric damage, implying that the mouse model of gastritis has been successfully established.
Plasma extravasation is widely acknowledged as a pivotal indicator of neurogenic inflammation. Evan’s blue, which exhibits a strong affinity for serum albumin, has been utilized to quantify the extent of extravasation accompanying inflammation [25]. To investigate whether visceral pain, such as that induced by GU, can be reflected on the body surface, causing a neurogenic inflammatory response, EB was administered via injection into the lateral caudal vein after GU modeling to quantify the extent of plasma extravasation. As shown in Figure 1C–J, the results suggested that the points of EB extravasation were predominantly distributed in the upper-back regions spanning the T9 to T11 segments, and at the operative incision in the GU mice, rather than in the control mice.
Moreover, to determine the referred mechanical hypersensitivity responses triggered by neurogenic inflammation in GU mice, von Frey filaments were employed to evaluate mechanical thresholds in the upper-back exudation points across the T9 to T11 segments. As illustrated in Figure 1K, the mechanical thresholds in the T9–T11 upper back exduation points of GU mice were lower than those in the control group (p < 0.0001), suggesting that the gastric ulcer induces somatosensory nociceptive sensitization in the upper back exudation points from the T9–T11 segments. Therefore, the upper back cutaneous exduation points from the T9–T11 segments are recognized as the sensitized points.
3.2. Acetic Acid-Induced Inflammation Increases the T-Type Ca2+ Currents (IT-Type) in DRG Neurons with Small Diameters but Not Medium Diameters
Based on the classical theory of DRR and AR, visceral inflammatory pain can affect cutaneous hypersensitivity through neurogenic inflammation, which is triggered by abnormal discharges of nociceptive neurons in DRGs that innervate the visceral organs [26]. Voltage-gated Ca^2+^ channels are critical for the active electrical properties of neurons, which are essential for propagating and modulating pain signals. In mice, DRG neurons are categorized as large- (>30 μm), medium- (20–30 μm), or small- (<20 μm) diameter [6]. DRG neurons with somatic diameters less than 30 μm are considered nociceptors as they can detect noxious stimuli and initiate pain responses [27]. To evaluate the differences in total calcium currents in T9–T11 small- and medium-diameter DRG neurons between control and GU mice, we employed a combination of voltage gating and pharmacological recording methods (Figure S1A). The membrane capacitances of different diameters DRG neurons are presented in Supplementary Table S1. As illustrated in Figure S1B, the ICa current densities (p < 0.05) and peak amplitude of ICa current (p < 0.01) in small-diameter DRG neurons from the GU group were markedly greater than those observed in the control group. Conversely, there are no significant differences in ICa current densities and peak amplitude of ICa current in the medium-diameter DRG neurons between the two groups (Figure S1D,E). These findings indicate that only the ICa currents of small-diameter DRG neurons exhibit significant changes in the GU group, potentially contributing to the action potential (AP) initiation and propagation, thereby further altering the neuronal excitability.
T-type Ca^2+^ channels have the distinctive capacity to become activated after a minor cellular membrane depolarization. This characteristic enables T-type channels to function close to the resting membrane potential, thereby influencing neuronal excitability [12]. Then, to ascertain whether the increased total ICa current density observed in the small-diameter DRG neurons is predominantly mediated by T-type Ca^2+^ channels, we first evaluated the changes in the T-type channel expression of T9–T11 DRGs. The immunofluorescent staining revealed a significant enhancement in the number of Ca_v_3.2-positive DRG neurons in GU mice compared to control mice (Figure 2A, p < 0.001).
In addition to observing the expression of the T-type channel changes, we further isolated and recorded IT-type (Figure 2B,C). The membrane capacitances of different diameters of DRG neurons are presented in Table 1. As shown in Figure 2D, patch-clamp recordings revealed significantly higher IT-type densities in small-diameter DRG neurons of GU mice than those in the control group (p < 0.01). In these small-diameter DRG neurons of the control group, the V1/2 of T-type channel current was −18.78 ± 2.32 mV and the k was 2.33 ± 1.01, respectively, while in the GU group, the V1/2 was −29.27 ± 2.93 mV (t = 2.323, p = 0.0426, unpaired t-test) and the k was 1.43 ± 0.27 (t = 1.008, p = 0.3352, unpaired t-test). The activation curve of T-type current fitted by a Boltzmann function exhibited a 10.49 mV hyperpolarizing shift in the small-diameter DRG neurons of GU mice (Figure 2F, p < 0.05). Subsequently, the steady-state inactivation curve showed no significant differences in the small-diameter DRG neurons between control (V1/2 = −87.49 ± 4.81 mV, k = −14.42 ± 2.61) and GU (V1/2 = −84.73 ± 4.42 mV, t = 0.4220, p = 0.6771, unpaired t-test; k = −13.25 ± 3.54, t = 0.2664, p = 0.7924, unpaired t-test) groups (Figure 2G). Moreover, there was no obvious change in the I_T-type_ densities of the medium-diameter DRG neurons between the two groups (Figure 2E). The V1/2 and k of the T-type activation curves in the medium-diameter DRG neurons between control (V1/2 = −29.52 ± 2.897 mV, k = 3.50 ± 1.05) and GU (V1/2 = −29.74 ± 2.669 mV, t = 0.047, p = 0.9629, unpaired t-test; k = 1.03 ± 0.32, t = 1.906, p = 0.0759, unpaired t-test) mice showed no obvious differences (Figure 2H). The steady-state inactivation curve showed no obvious differences in the medium-diameter DRG neurons between control (V_1/2_ = −81.80 ± 5.52 mV, k = −14.54 ± 4.26) and GU (V_1/2_ = −82.00 ± 5.04 mV, t = 0.0274, p = 0.9784, unpaired t-test; k = −5.83 ± 0.97, t = 2.075, p = 0.0505, unpaired t-test) groups (Figure 2I). These results suggest that both the gating properties and expression of T-type channel changes may contribute to the alterations of total I_Ca_ current.
3.3. Inhibition of the Chemokine CCR2 Reduces IT-Type in Small-Diameter DRG Neurons and Attenuates Somatic Neurogenic Inflammation and Referred Mechanical Hypersensitivity in Gastric Ulcer Mice
CCR2 is the main receptor for CCL2 released from DRG neurons [28] and directly excites these nociceptive neurons by influencing their ion channels [6]. Previous research has shown that the upregulation of CCR2 on DRG is vital for pain induction, while CCR2 antagonists can induce analgesia [14]. Therefore, to clarify whether the alteration of T-type Ca^2+^ channel currents in the GU mice was mediated by the activation of CCR2 on T9–T11 DRG, we first evaluated the co-expression of CCR2 with Ca_v_3.2 within the T9–T11 DRG neurons in two groups. Immunofluorescent staining revealed a significant enhancement in co-labeled CCR2 and Ca_v_3.2-positive DRG neurons in the GU mice compared to the control group (Figure 3A, p < 0.001). Except for observing changes in CCR2 expression, we further investigated the role of CCR2 in modulating T-type Ca^2+^ channel currents by applying the CCR2 antagonist (RS102895,10 μM) to small-diameter DRG neurons in the GU group. The membrane capacitances of these neurons are presented in Table 1. The results indicated that the current densities of T-type Ca^2+^ channels in small-diameter DRG neurons of the RS102895 group were markedly decreased in comparison to those in the vehicle group (Figure 3E, p < 0.001). In the small-diameter DRG neurons of the vehicle group, the V1/2 and k were −29.27 ± 2.93 mV and 1.43 ± 0.27, respectively, while after washing the RS102895, the values became to −20.13 ± 1.06 mV (t = 2.588, p = 0.0237, unpaired t-test) and 1.30 ± 0.24 (t = 0.3380, p = 0.7404, unpaired t-test), respectively. The activation curve of T-type Ca^2+^ channel currents fitted by a Boltzmann function exhibited a notable 9.14 mV depolarizing shift in the small-diameter DRG neurons after treatment with RS102895 (Figure 3F, p < 0.05). Moreover, as illustrated in Figure 3G, the steady-state inactivation curve showed no significant difference in the small-diameter DRG neurons between the vehicle and RS102895 groups. These findings imply that the augmented T-type I_Ca_ in small-diameter DRG neurons in GU mice may be mediated by the activation of CCR2 and that its antagonists can effectively reduce this activation.
Then, we investigated the effect of RS102895 (5 mg/kg), the CCR2 antagonist, on the number of EB exudate points and nociceptive responses in the GU mice. As illustrated in Figure 3, RS102895 markedly reduced the number of EB exudate points (Figure 3H, p < 0.0001) and enhanced the mechanical pain threshold (Figure 3I, p < 0.0001), thereby alleviating referred mechanical hypersensitivity in the somatic areas, particularly in the upper-back regions from T9 to T11 segments in GU mice compared to the vehicle group. These findings demonstrate that CCR2 is essential in the somatosensory nociceptive sensitization induced by viscera pain, potentially by mediating the gating properties of T-type channels in the small-diameter DRG neurons.
The above results highlight the essential relationship between neurogenic inflammation and referred mechanical hypersensitivity in somatic regions and the upregulation of CCR2 expression induced by visceral pain. Nevertheless, it remains unclear if activating CCR2 in the small-diameter DRG neurons of control mice can partially mimic the alterations in the gating properties of T-type channels associated with visceral pain. To investigate this, we assessed the I_T-type_ in T9–T11 small-diameter DRG neurons of control mice treated with CCR2 agonist CCL2 (100 nM) during patch-clamp recordings. The membrane capacitances of these neurons are presented in Table 1. As illustrated in Figure S2, the T-type current density significantly increased with a hyperpolarizing activation curve shift underlying the role of exogenous CCL2 (p < 0.01). In the small-diameter DRG neurons of the vehicle group, the values of V1/2 and k of the T-type channel currents were −18.78 ± 2.32 mV and 2.33 ± 1.01, respectively, whereas after washing the CCL2, the values shifted to −27.50 ± 1.85 mV (t = 2.948, p = 0.0185, unpaired t-test) and 1.99 ± 0.52 (t = 0.3234, p = 0.7524, unpaired t-test). The activation curve of T-type Ca^2+^ channel currents, fitted by a Boltzmann function, exhibited a hyperpolarizing shift of approximately 8.72 mV in the small-diameter DRG neurons (Figure S2E, p < 0.05). These results suggest that exogenous CCL2 interacts with CCR2 in small-diameter DRG neurons of naive mice, potentially mimicking the effects of endogenous CCL2 acting on CCR2 in small-diameter DRG neurons of GU mice. The underlying mechanisms may involve the activation and upregulation of CCR2, which alters the dynamics of T-type channels.
4. Discussion
Although ion channels and chemokines in DRG neurons are known to participate in the pathogenesis of visceral pain, the distinct changes and regulatory mechanisms of T-type Ca^2+^ channel and CCR2 in different-sized DRG neurons after visceral injury are not well understood. Our study explored the underlying ion channel mechanisms of CCR2 on DRG neurons using acetic acid to construct a visceral pain model. We found that CCL2 directly increases T-type Ca^2+^ currents in normal small-diameter DRG neurons, which mimics the effects of endogenous CCL2 acting on CCR2, inducing the enhancement of T-type channel currents in GU mice. On the contrary, the administration of RS102895, a blocker of CCR2, reduced acetic acid-mediated referred somatic sensitization and T-type channel currents in small-diameter DRG neurons, which were distributed on the sensitized somatic area. These findings collectively indicated that the upregulation of CCR2 and T-type channel expressions, enhancing T-type channel currents in small-diameter DRG neurons, contributed to visceral pain-induced referred somatic hyperalgesia.
Visceral lesions may manifest as neurogenic inflammatory responses on specific somatic regions. Numerous studies have demonstrated that multiple EB extravasation points appear on the skin overlying the dorsal T6–T11 spinal cord segments in rat models of gastric injury, including minor curvature ulcers, acute and chronic gastric ulceration, and gastritis [29,30,31,32]. The quantity and area of EB extravasation points increase proportionally with the severity of gastric injury in mice [33]. These observations suggest that EB extravasation points are indicators of visceral pain-induced neurogenic inflammation, reflecting the cutaneous areas implicated in visceral pain disorders. In this study, using EB and von Frey tests, we noticed that in mice treated with acetic acid, the gastric referred area was primarily found on the upper back, innervated by T9–T11 segments, and within the gastric innervation. This neurogenic inflammation of GU mice is primarily driven by alterations in the functional activity of primary sensory neurons, particularly DRG neurons, leading to a lowered activation threshold and the heightened excitability of peripheral nerve endings that project to the affected areas, thereby resulting in the sensitization of referred somatic areas.
Referred somatic hyperalgesia is linked to the hyperexcitability of DRG neurons [34]. When the nervous system detects changes in the body’s internal environment, the membrane potential of sensory neurons may attain the threshold potential level, eliciting the generation of action potentials that encode corresponding sensory information in strings. Therefore, the alterations in the neuronal excitatory state constitute a critical prerequisite for sensory information transmission. Several studies highlighted the significant role of T-type Ca^2+^ channels in elevating the nociceptive neuron excitability by lowering the AP firing threshold and enhancing Ca^2+^ entry during AP propagation [35]. In this study, we concentrated on the small-diameter DRG neurons since they are identified as nociceptive neurons [6]. The results displayed that the mean values of T-type current density are 20.29 ± 16.20 pA/pF in the control and 75.96 ± 9.803 pA/pF in the model group at −20 mV, respectively, with CsCl internal solution. These findings diverge from those of other laboratories. For instance, a study, using the KCl internal solution, reported the T-type current density values of the small-sized (<25 pF) IB4-negative DRG neurons below 40 pA/PF at −20 mV in both control and model rats [36]. However, another one demonstrated the T-type current density values of the small-sized (<30 μm diameter) IB4-positive DRG neurons in model rats were markedly higher (76.66 ± 5.62 pA/pF) than control rats (43.33 ± 8.19 pA/pF) at −30 mV, utilizing a tetramethylammonium hydroxide internal solution [37]. Another study in rats, utilizing a Cs-methanesulfonate internal solution, recorded average peak T-type currents density values of 25 ± 2 pA/pF in normal and 93 ± 8 pA/pF in T-rich IB4-positive small-diameter (26–31 μm) DRG neurons at −20 mV [38]. Compared with these studies, such discrepancies may be attributed to a variety of factors, including the different DRG segments, electrophysiological recording conditions (e.g., temperatures, internal solutions, and voltage protocols), cell types (peptidergic or non-peptidergic), and species-specific differences in Ca_v_3 (T-type) isoform expression between mice and rats, as well as age-related differences influencing neuronal activity and T-type current densities. In brief, our findings revealed notable increases in both the current density and expression of T-type Ca^2+^ channels in small-diameter DRG neurons, whereas no such changes were detected in medium-diameter DRG neurons in GU mice. These enhancements may contribute to the hyperexcitability of small-diameter DRG neurons following referred visceral pain, thereby promoting the releases of the neuropeptides such as calcitonin-gene-related peptide (CGRP), histamine (HA), and substance P to referred sensitized areas, which can induce the vasodilation and extravasation of plasma proteins, resulting in referred somatic pain [39]. The functional alterations of T-type channel current in small DRG neurons may modulate their neuronal excitabilities and AP generations which is paramount to comprehending their pathophysiological significance in referred somatic pain models. Hence, future investigations should explore the AP threshold (rheobase), firing frequency, and additional membrane properties, which collectively reflect the intrinsic excitability of small-diameter DRG neurons.
CCR2 is the principal receptor for CCL2, critical for recruiting pro-inflammatory monocytes to inflamed tissue [40]. The release of CCL2 can enhance the excitability of DRG neurons, thereby facilitating pain sensitization [41,42]. In alignment with the proalgesic action of CCL2, the knockout of CCR2 has been shown to alleviate inflammatory and neuropathic pain resulting from chronic injury [16,17,18]. Additionally, both the short-term blockade and depletion of Ca_v_3.2 have also demonstrated analgesic effects. Moreover, T-type Ca^2+^ channels can be inhibited by CCR2 antagonists [43]. Therefore, it is plausible that a combined Ca_v_3.2 and CCR2 antagonist may exhibit synergistic effects in pain treatment. Consistent with these findings, our data showed a large increase in DRG neurons co-labeled with CCR2 and Ca_v_3.2 in GU mice. The current densities of the T-type Ca^2+^ channels were significantly decreased, coinciding with a reduction in the number of EB extravasation points and the alleviation of referred mechanical hypersensitivity in GU mice following the administration of RS102895. Nevertheless, the potential possibility for the intraperitoneal injection of a CCR2 inhibitor to induce other adverse effects, alongside the necessity for long-term pharmacokinetic analysis in mice, requires further investigation. Meanwhile, in future studies, the application of various chemokine receptor antagonists to verify target specificity in referred hyperalgesia arising from visceral pain would be crucial in achieving a more profound and comprehensive understanding of this phenomenon. Additionally, exposure to CCL2 in normal small-diameter neurons increased the T-type Ca^2+^ channel currents to levels comparable to those observed in GU mice. Hence, the activation and upregulation of CCR2 appear to alter the gating properties of T-type Ca^2+^ channels, potentially serving as underlying mechanisms for referred somatic sensitization during the visceral pain states. Further investigation is warranted to determine whether the effects of CCL2 on DRG neurons with small diameters act directly on CCR2 receptors, subsequently influencing the gating properties of T-type Ca^2+^ channels. In our research, we exclusively utilized the patch-clamp method to confirm the regulatory effect of CCR2 on T-type Ca^2+^ channels. Further exploration utilizing a wider array of experimental methods is necessary to elucidate the specific mechanisms of interaction between CCR2 and T-type Ca^2+^ channels. Whether CCR2 antagonist can exert long-term effects is also a key focus of our future research. Additionally, it remains to be established whether the administration of CCL2 to naive mice can induce somatic hyperalgesia.
The limitations of our study include the following points: (1) We currently categorize nociceptive neurons within DRG cell populations based on soma size and average capacitance; however, these approaches prove insufficient for a comprehensive characterization. Given these limitations and inadequacies, future studies are necessary to ascertain whether these small-diameter DRG neurons involved in the referred pain exhibit peptidergic or non-peptidergic properties. Such studies should integrate molecular profiling (e.g., IB4, TRPV1, CGRP) to more precisely define neuronal subtypes. For instance, IB4-positive neurons typically correspond to non-peptidergic C-fibers, whereas TRPV1- or CGRP-positive populations are linked to peptidergic nociceptive pathways [36]. These markers will enhance cross-species comparisons, particularly addressing size discrepancies in homologous phenotypes (e.g., rat DRG neurons with analogous functions). (2) Considering in vitro experiments, such as the patch-clamp technique employed in our work, cannot directly elucidate the causal relationship between hyperalgesia and DRG neuronal activities nor the specific mechanisms underlying the interaction between CCR2 and T-type Ca^2+^ channels, it is essential that future studies incorporate more sophisticated in vivo methodologies, such as calcium imaging, single-cell sequencing, co-immunoprecipitation, and cryo-electron microscopy to enhance our understanding.
5. Conclusions
The present study demonstrates that the activation of T-type Ca^2+^ channels in small-diameter DRG neurons is associated with GU-related neurogenic plasma extravasation and referred somatic allodynia. CCR2 may act as a regulator of neurogenic inflammation and mechanical allodynia by modulating the expression and gating properties of the T-type Ca^2+^ channels. The inhibitor of CCR2 may be a useful and novel therapeutic tool for visceral pain treatment.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Jin Q. Chang Y. Lu C. Chen L. Wang Y. Referred pain: Characteristics, possible mechanisms, and clinical management Front. Neurol.202314110481710.3389/fneur.2023.110481737448749 PMC 10338069 · doi ↗ · pubmed ↗
- 2Gebhart G.F. Bielefeldt K. Physiology of Visceral Pain Compr. Physiol.201661609163310.1002/cphy.c 15004927783853 · doi ↗ · pubmed ↗
- 3Li D. Ren Y. Xu X. Zou X. Fang L. Lin Q. Sensitization of primary afferent nociceptors induced by intradermal capsaicin involves the peripheral release of calcitonin gene-related Peptide driven by dorsal root reflexes J. Pain 200891155116810.1016/j.jpain.2008.06.01118701354 PMC 2642671 · doi ↗ · pubmed ↗
- 4Hagains C.E. Trevino L.A. He J.W. Liu H. Peng Y.B. Contributions of dorsal root reflex and axonal reflex to formalin-induced inflammation Brain Res.20101359909710.1016/j.brainres.2010.08.09720816764 · doi ↗ · pubmed ↗
- 5Abd-Elsayed A. Vardhan S. Aggarwal A. Vardhan M. Diwan S.A. Mechanisms of Action of Dorsal Root Ganglion Stimulation Int. J. Mol. Sci.202425359110.3390/ijms 2507359138612402 PMC 11011701 · doi ↗ · pubmed ↗
- 6Li L. Liu Y. Hu W. Yang J. Ma S. Tian Z. Cao Z. Pan K. Jiang M. Liu X. Peripheral CCL 2 induces inflammatory pain via the regulation of Ih currents in small-diameter DRG neurons Front. Mol. Neurosci.202316114461410.3389/fnmol.2023.114461437860084 PMC 10582564 · doi ↗ · pubmed ↗
- 7Hu S. Xiao Y. Zhu L. Li L. Hu C.Y. Jiang X. Xu G.Y. Neonatal maternal deprivation sensitizes voltage-gated sodium channel currents in colon-specific dorsal root ganglion neurons in rats Am. J. Physiol. Gastrointest. Liver Physiol.2013304 G 311G 32110.1152/ajpgi.00338.201223139220 · doi ↗ · pubmed ↗
- 8Michel N. Narayanan P. Shomroni O. Schmidt M. Maturational Changes in Mouse Cutaneous Touch and Piezo 2-Mediated Mechanotransduction Cell Rep.20203210791210.1016/j.celrep.2020.10791232697985 · doi ↗ · pubmed ↗