Lysosomes’ fallback strategies: more than just survival or death
Quan Wang, Ruolin Wang, Haihui Hu, Xiaoqing Huo, Fulong Wang

TL;DR
This paper reviews how lysosomes respond to damage and how these responses contribute to diseases like neurodegeneration and cancer.
Contribution
The paper highlights alternative lysosomal strategies beyond repair, linking them to disease progression and treatment resistance.
Findings
Lysosomal secretion of contents and interactions with other organelles are key responses to damage.
Lysosomal fallback strategies are linked to neurodegenerative diseases and cancer drug resistance.
Organelles like the ER and Golgi play auxiliary roles in lysosomal damage responses.
Abstract
Lysosomes are heterogeneous, acidic organelles whose proper functionality is critically dependent on maintaining the integrity of their membranes and the acidity within their lumen. When subjected to stress, the lysosomal membrane can become permeabilized, posing a significant risk to the organelle’s survival and necessitating prompt repair. Although numerous mechanisms for lysosomal repair have been identified in recent years, the progression of lysosome-related diseases is more closely linked to the organelle’s alternative strategies when repair mechanisms fail, particularly in the contexts of aging and pathogen infection. This review explores lysosomal responses to damage, including the secretion of lysosomal contents and the interactions with lysosome-associated organelles in the endolysosomal system. Furthermore, it examines the role of organelles outside this system, such as the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
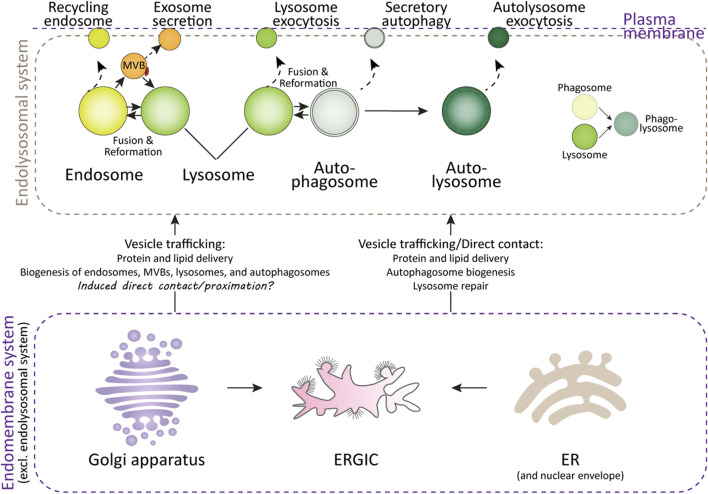 FIGURE 1
FIGURE 1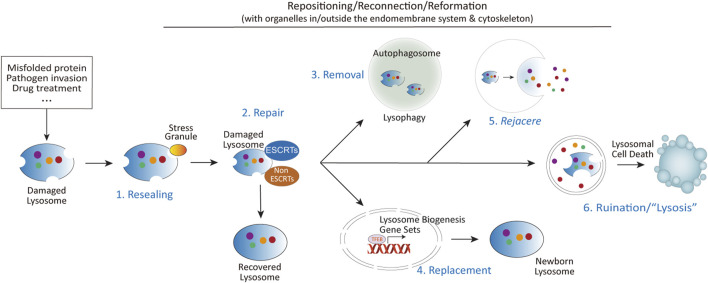 FIGURE 2
FIGURE 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCalcium signaling and nucleotide metabolism · Autophagy in Disease and Therapy · Lysosomal Storage Disorders Research
Introduction
Lysosomes are the primary degradation organelles of the cell and serve as the terminal compartments within the endolysosomal system (Ballabio and Bonifacino, 2020). This system comprises a network of dynamic and interconnected organelles, including endosomes, multivesicular bodies (MVBs), lysosomes, lysosome-related organelles (e.g., granules in immune cells), phagosomes, and autophagosomes (Figure 1). From a spatiotemporal perspective, rather than functioning as isolated compartments, the endolysosomal system operates as a highly coordinated and adaptive “digestive tract,” facilitating the sequestration, sorting, and degradation of cargo derived from both intracellular and extracellular sources (Hu et al., 2015; Neel et al., 2024). Endosomes, phagosomes, and autophagosomes act as intermediary hubs within this network, directing cargo toward lysosomes, which serve as the terminal degradative compartments where cellular digestion and recycling occur. Beyond their degradative role, components of this system are involved in specialized secretion pathways (Figure 1): autophagosomes facilitate secretory autophagy, MVBs mediate the release of exosomes, and lysosomes participate in lysosome-associated exocytosis, thereby expelling undigested material, especially when the degradation function of the lysosomes is compromised (Buratta et al., 2020; Han et al., 2022; Ponpuak et al., 2015).
Lysosomes and their interacting organelles. Top panel: Overview of lysosomes and their interacting organelles within the endolysosomal system. Lysosomal fusion and fission with endosomes, autophagosomes, and phagosomes are critical for their maturation, functional maintenance, reformation, and repair after damage. However, when lysosomal damage exceeds the capacity for repair, both lysosomes and the organelles within the endolysosomal system may undergo secretion in various forms. Bottom panel: Interaction of lysosomes with organelles in the endomembrane system. The delivery of proteins and lipids from the ER and Golgi apparatus is essential for lysosomal function and repair. Additionally, the ER, ER-Golgi intermediate compartment (ERGIC), and Golgi apparatus contribute to autophagosome formation, a process critical for the clearance of damaged lysosomes.
From an endolysosome-centric perspective, other organelles within the broader endomembrane system—such as the ER, Golgi apparatus, and plasma membrane—serve as auxiliary organelles or components of the endolysosomal system (Figure 1). These organelles support lysosomal function by synthesizing and delivering lipids, proteins, and membranes via vesicular trafficking, membrane budding, and direct membrane contact sites. These interactions ensure the proper functioning of the endolysosomal system and maintain cellular homeostasis (De Tito et al., 2020; Nascimbeni et al., 2017; Sun et al., 2024; Tan and Finkel, 2023).
Therefore, when lysosomes are exposed to various stresses and their membrane integrity is compromised, organelles within the endolysosomal and endomembrane systems may play critical roles in facilitating lysosomal repair. These stresses can arise from physiological, pathological, and external factors that impair membrane integrity and enzymatic function. Physiologically, aging contributes to lysosomal damage by promoting the accumulation of indigestible materials, such as lipofuscin, which reduces degradation efficiency, while oxidative stress induces lipid peroxidation within lysosomal membranes, weakening their structure and leading to enzyme leakage (Gahlot et al., 2024; Pan et al., 2021; Zhang et al., 2023). Pathological conditions—including neurodegenerative diseases (Root et al., 2021), lysosomal storage disorders (Parenti et al., 2021), metabolic disturbances (Almeida et al., 2020), and infections (Richards et al., 2022)—exacerbate these effects by causing substrate overload, protein aggregation, and inflammation, further destabilizing lysosomes. Additionally, external agents such as lysosomotropic drugs, ionophores, toxins, and environmental pollutants can directly damage lysosomal membranes, triggering the excessive release of lysosomal enzymes. In research settings, compounds like L-leucyl-L-leucine methyl ester (LLOMe), Glycyl-L-phenylalanine 2-naphthylamide (GPN), methyl-serine dodecylqamide hydrochloride (MSDH), chloroquine, Bafilomycin A1, ammonium chloride, silica crystals, and overexpression of mutant proteins associated with neurodegenerative diseases and lysosomal storage disorders—such as alpha-synuclein, amyloid-beta, Tau, Huntingtin, TDP43, SOD1, PANK2, NPC1, NPC2, CLN3, GBA1, HEXA, GAA, MPS, ASAH1, CTNS, and GALC—are commonly used to mimic these conditions (Parenti et al., 2021; Root et al., 2021; Udayar et al., 2022). Pathogen-derived factors, such as SapM, PtpA, and ESAT-6 from Mycobacterium tuberculosis (Ramon-Luing et al., 2023); Listeriolysin O (LLO) from Listeria monocytogenes (Shaughnessy et al., 2006); SopB from Salmonella enterica (Bakowski et al., 2010); Nef from HIV (Sanfridson et al., 1997); and ORF3a from SARS-CoV-2 (Walia et al., 2024), are also employed to study lysosomal dysfunction.
To date, a wide array of key proteins and complexes have been identified as being recruited to the damaged lysosomal sites to mediate repair processes. These include the Endosomal Sorting Complexes Required for Transport (ESCRT) machinery (Jia et al., 2020b; Radulovic et al., 2018; Skowyra et al., 2018), Annexins (Ebstrup et al., 2023; Yim et al., 2022), mTOR (Jia et al., 2018; Jia et al., 2019), AMPK (Jia et al., 2020a), PI4K2A (Tan and Finkel, 2022), and stress granules (Bussi et al., 2023; Duran et al., 2024; Jia et al., 2022). However, the successful execution of lysosomal repair or resolution of lysosomal stress is heavily dependent on the collaboration and interactions with other organelles. This includes fusion and fission events involving lysosomes, endosomes, and autophagosomes (Bhattacharya et al., 2023; Maejima et al., 2013; Rodgers et al., 2022; Saffi and Botelho, 2019), as well as both direct and indirect interactions with the ER (Radulovic et al., 2022; Tan and Finkel, 2022; Wang et al., 2024), Golgi apparatus (Lie et al., 2021; Vitry et al., 2010), and plasma membrane (Domingues et al., 2024; Sho et al., 2024; Wang et al., 2023; Zhong et al., 2023) (Figure 1).
The progression of lysosome-related diseases often correlates more closely with alternative strategies employed by lysosomes when conventional repair mechanisms fail, particularly in the contexts of age-related diseases and pathogen infections. This review aims to critically examine these alternative strategies, including different forms of lysosomal damage-related secretion, and the role ER and Golgi aparatus play in the repair and handling of damged lysosomes—other important aspects related to lysosomal damage repair, such as lysosomal repositioning and reformation, and lysosomal cell death (Figure 2) are reviewed elsewhere (Aits and Jäättelä, 2013; Gómez-Sintes et al., 2016; Pu et al., 2016; Scerra et al., 2022; Wang et al., 2018), therefore would not be covered here. By addressing these relatively neglected areas of lysosomal adaptation and alterations following lysosomal damage, this review seeks to provide a better understanding of the underlying mechanisms and their implications for cellular homeostasis and disease.
The diverse fates of damaged lysosomes. Upon lysosomal damage caused by aggregated proteins, invading pathogens, chemical insults, or other stressors, several outcomes are possible: (1) Immediate resealing of lysosomal membranes through stress granule-mediated repair. (2) Activation of multiple repair mechanisms, including ESCRTs dependent and independent mechanisms. (3) Mildly damaged lysosomes can be repaired, while severely damaged lysosomes are targeted for removal via autophagy (lysophagy). (4) Lysosomal damage also promotes lysosomal biogenesis through the activation of TFEB, regulated by mTORC1, calcium signaling, and other pathways. (5) In cases where repair is insufficient, damaged lysosomes may be expelled through fusion with the plasma membrane. The term rejacere (Latin: to expel) is used here to describe the active expulsion of lysosomal contents, emphasizing its mechanistic and evolutionary context. (6) If repair and removal mechanisms fail, the release of lysosomal contents such as cathepsins, coupled with altered signaling pathways at the lysosomal signaling hub, can trigger lysosomal cell death via multiple mechanisms.
Lysosomal damage and secretion
Under pathological conditions where lysosomes are severely damaged—such as during cancer treatment, pathogen infections, and neurodegenerative diseases—their repair capacities are often compromised or overwhelmed. In these scenarios, cells frequently adopt an alternative mechanism of expelling compromised lysosomes along with their enzymatic content and undigested materials (Chen et al., 2021; Domingues et al., 2024; Sho et al., 2024; Wang et al., 2023; Zhitomirsky and Assaraf, 2017; Zhong et al., 2023). These expelled materials may include lysosomotropic agents, engulfed pathogens, and aggregated pathogenic proteins. Furthermore, vesicles and organelles within the endolysosomal system that are primed to fuse with lysosomes—such as autophagosomes, endosomes, and MVBs—as well as damaged organelles engulfed by autophagosomes or endosomes [e.g., mitochondria (Bao et al., 2022; Liang et al., 2023)], may also be secreted concurrently or in parallel with lysosomal expulsion.
These lysosome-associated secretion processes can significantly influence disease susceptibility and progression (Groth-Pedersen and Jaattela, 2013; Scotto Rosato et al., 2022; Xu et al., 2021). For instance, the expulsion of aggregated proteins or pathogens through this mechanism has been linked to the spread of pathogenic agents in neurodegenerative diseases and the survival of invasive pathogens in infections. In cancer, lysosomal exocytosis contributes to drug resistance by releasing lysosomotropic chemotherapeutic agents from cells (Machado et al., 2015; Zhitomirsky and Assaraf, 2017). Conversely, under specific circumstances, lysosomal secretion may serve a protective role, such as by removing cytotoxic material or reducing intracellular stress (Tsunemi et al., 2019; Zhong et al., 2023).
The impact of lysosome-associated secretion on diseases underscores a potentially underappreciated relationship between this process and disease mechanisms. By better understanding the molecular pathways governing lysosomal secretion, therapeutic targeting of lysosome-associated secretion could represent a novel and promising strategy for managing diseases characterized by lysosomal dysfunction, including neurodegenerative disorders, infectious diseases, and cancers.
Lysosomal exocytosis
Lysosomal exocytosis is a calcium-dependent process in which lysosomes fuse directly with the plasma membrane to release their contents into the extracellular space (Zhong et al., 2023). This mechanism is present in all cell types and is activated by a variety of stimuli. It plays a crucial role in numerous physiological processes, including plasma membrane repair (Reddy et al., 2001), bone resorption (Kim et al., 2024; Lee et al., 2024), pigmentation (Stinchcombe et al., 2004; Wünkhaus et al., 2024), immune responses (Brady et al., 2018; Jin et al., 2021; Sáez et al., 2019), mitosis (Hämälistö et al., 2020; Nugues et al., 2022), and ATP release in the nervous system (Jung et al., 2013; Shin et al., 2012; Zhang et al., 2007). Moreover, when lysosomes are damaged or their functions are compromised, the exocytosis process is typically enhanced (Bogacki et al., 2025; Chen et al., 2011; Domingues et al., 2024; Eriksson et al., 2020; Ghosh et al., 2020; Sivaramakrishnan et al., 2012; Wang et al., 2023). This enhancement is triggered by events such as lysosomal membrane permeabilization, alkalinization, and the consequent release of Ca^2+^, all of which can induce cellular stress and inflammation.
Enhanced lysosomal exocytosis can have both beneficial and detrimental effects, depending on the pathological context. For instance, in cancer treatment, the release of lysosomal enzymes through lysosomal exocytosis is often associated with increased metastasis and reduced patient survival (Machado et al., 2015; Ren et al., 2025). Conversely, the expulsion of lysosomotropic agents via lysosomal exocytosis in renal proximal tubular epithelial cells—mediated by the activation of the lysosomal Ca^2+^ channel TRPML1—has been shown to mitigate uranium-induced nephrotoxicity (Zhong et al., 2023). This dichotomy underscores the critical influence of the specific components released during lysosomal exocytosis.
In neurodegenerative diseases, the accumulation of aggregated misfolded proteins is a central factor in disease progression. The role of lysosomes in degrading these proteins—or alternatively, in their activation, accumulation, or release—is paramount. For example, in synucleinopathy models, lysosomal exocytosis-mediated release of degradation-resistant α-synuclein species from neurons has been identified as a key mechanism for the propagation of pathogenic α-synuclein in mouse brains (Xie et al., 2022). However, contrasting findings indicate that activation of TRPML1 can protect human dopaminergic neurons by rescuing defective α-synuclein secretion and preventing its accumulation (Tsunemi et al., 2019). This again suggests that the effects of lysosomal exocytosis in neurodegenerative diseases may vary depending on the specific cellular context involved.
Similarly, during pathogen infections, lysosomal damage-induced exocytosis serves as an effective mechanism to expel overwhelmed pathogens (Chen et al., 2021; Deretic and Wang, 2023; Ghosh et al., 2020; Koo et al., 2008; Shtuhin-Rahav et al., 2023; Wang et al., 2023). However, this process can be hijacked by pathogens to facilitate their own survival and dissemination. For example, both severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (Chen et al., 2021) and its β-coronavirus relative MHV (Ghosh et al., 2020) induce lysosomal damage and exocytosis through multiple Ca^2+^-dependent mechanisms, enabling their egress from infected cells. Additionally, in bacterial infections, lysosomal exocytosis from immune and other cell types is associated with the cytolytic effects of bacteria, allowing bacteria within lysosomes and phagosomes to evade digestion and enhance their survival (Koo et al., 2008; Shtuhin-Rahav et al., 2023; Wang et al., 2023).
These studies illustrate that lysosomal exocytosis plays a complex role in disease dynamics, acting as a double-edged sword. On one hand, it can protect cells by removing toxic substances and pathogens; on the other hand, it can facilitate disease progression by promoting metastasis, protein propagation, and pathogen survival. Understanding the specific context of lysosomal exocytosis is therefore crucial.
Alternative secretion mechanisms
Besides lysosomal exocytosis, at least two additional secretion mechanisms are linked to lysosomal damage and compromised lysosomal function: exosome secretion and secretory autophagy. Exosome secretion is a process in which small extracellular vesicles, known as exosomes, are released following the fusion of MVBs with the plasma membrane (Han et al., 2022). The formation of exosomes is facilitated by the ESCRT complexes, which sort cargo into intraluminal vesicles and drive membrane inward budding events. These exosomes, which transport proteins, lipids, and nucleic acids, play a crucial role in intercellular communication and modulate various physiological and pathological processes. Secretory autophagy, on the other hand, repurposes the cellular autophagic machinery to facilitate the secretion of cellular components, rather than their degradation (Buratta et al., 2020; Debnath and Leidal, 2022; Solvik et al., 2022). Unlike the classical autophagic pathway, which typically directs cargo to lysosomes for degradation, secretory autophagy allows the release of cellular materials, such as proteins and lipids, into the extracellular space. This process is particularly important for cellular communication, immune responses, and tissue homeostasis, providing an alternative route for the secretion of factors like cytokines and extracellular matrix components. Both exosome secretion and secretory autophagy serve as alternative pathways for managing cargo within the endolysosomal system, contributing to cellular responses and disease progression by facilitating the extracellular release of materials that would otherwise be degraded.
A significant feature of approximately half of the lysosomal storage disorders (LSDs)—a group of over 70 rare inherited metabolic diseases caused by lysosomal dysfunction—is the increased secretion of exosomes, highlighting the correlation between lysosomal impairment and exosome release (Abe et al., 2024; Alvarez-Erviti et al., 2011). The exosome-mediated release of pathogenic α-synuclein from macrophage lineage cells or neuroblastoma cells has been attributed to lysosomal stress-induced dysfunction (Abe et al., 2024; Alvarez-Erviti et al., 2011). In another model of lysosomal dysfunction, the contents released via exosomes include amyloid precursor protein C-terminal fragments (APP-CTFs), specific sphingolipids, and the phospholipid bis(monoacylglycero)phosphate (BMP), which normally reside in the internal vesicles of endolysosomes (Miranda et al., 2018). Notably, disruption of endolysosome fusion also increases exosome secretion (Shelke et al., 2023), indicating that the physical interaction between lysosomes and MVBs inherently predicts exosome release.
However, when lysosomal inhibition is induced by agents such as chloroquine or Bafilomycin A, cells tend to utilize secretory autophagy instead of exosome secretion, as evidenced by increased secretion of autophagy cargo receptors in extracellular vesicles and particles (EVPs) but negligible changes in classical exosome markers such as TSG101, ALIX, and CD9 (Solvik et al., 2022). This observation is consistent with several other studies demonstrating that lysosomal damage or stress can induce secretory autophagy (Chang et al., 2024; Dash et al., 2024; Kimura et al., 2017). Considering the significant roles of secretory autophagy and exosome secretion in the progression of diseases such as neurodegenerative disorders and cancer, as well as the use of exosomes for drug delivery, the interplay between lysosomal damage and secretion warrants further attention.
Lysosomal damage and endomembrane system
While much attention has been given to the endolysosomal system itself, emerging research reveals that lysosomes rely heavily on interactions with other organelles—particularly the endoplasmic reticulum (ER) and Golgi apparatus—to repair damage and restore homeostasis.
ER and lysosomal damage
Emerging evidence underscores the critical role of endoplasmic reticulum (ER)-lysosome membrane contact sites (MCSs) in mediating lysosomal repair. Central to this process is the phosphatidylinositol-4 kinase type 2α (PI4K2A), which generates phosphatidylinositol-4-phosphate (PI4P) at lysosomal membranes. PI4P recruits oxysterol-binding protein (OSBP)-related protein (ORP) family members, including ORP9, ORP10, ORP11, and OSBP, to orchestrate the formation of ER-lysosome MCSs (Tan and Finkel, 2022). These dynamic contact sites facilitate the transfer of phosphatidylserine and cholesterol from the ER to damaged lysosomes, enabling rapid membrane restoration. Complementary studies emphasize the role of cholesterol and ER-resident tethering proteins, such as VAPA/B and ORP1L, in stabilizing these interactions and promoting lysosomal integrity (Radulovic et al., 2022).
A parallel mechanism involves the lipid transfer protein ATG2, which is recruited to damaged lysosomes to mediate direct lipid transfer for membrane repair (Tan and Finkel, 2022). ATG2’s interaction with the lipid scramblase ATG9, essential for ER-phagophore contact site formation and autophagosome maturation (Gomez-Sanchez et al., 2018; van Vliet et al., 2022), suggests a coordinated interplay between lysosomal repair and lysophagy. This duality highlights a potential regulatory axis where ER-driven lipid redistribution supports both autophagosome biogenesis and lysosomal membrane restoration.
Pathophysiological insights emerge from studies linking ER-lysosome interactions to Parkinson’s disease (PD). VPS13C and LRRK2, two PD-associated proteins, are recruited to damaged lysosomes: VPS13C facilitates ER-lysosome tethering, while LRRK2 promotes lysosomal membrane tubulation and cargo sorting (Bonet-Ponce and Cookson, 2022; Wang et al., 2024). Similarly, PDZD8, a tubular lipid-binding protein (TULIP) superfamily member, tethers ER-lysosome MCSs to regulate lysosome maturation and autophagy (Guillén-Samander et al., 2019; Thakur and O’Connor-Giles, 2023). Behavioral abnormalities observed in PDZD8-deficient mice (Kurihara et al., 2023) raise intriguing questions about whether these phenotypes come partially from disrupted ER-lysosome communication, underscoring the need to explore MCS dysfunction in neurodegenerative contexts.
The endoplasmic reticulum (ER) may contribute to lysosomal damage repair through additional mechanisms. For example, during lysosomal injury, calcium efflux from damaged lysosomes has been shown to trigger compensatory ER-mediated calcium refilling in multiple models of lysosomal dysfunction (Garrity et al., 2016; Kang et al., 2024; Liu and Lieberman, 2019). Given the central role of calcium signaling in lysosomal repair and adaptation, future studies should investigate whether ER-lysosome membrane contact sites (MCSs) directly facilitate this calcium replenishment process. Such work could reveal how spatial and temporal coordination between calcium homeostasis and lipid transfer synergistically enhances lysosomal membrane repair. Beyond calcium dynamics, the ER and ER-Golgi intermediate compartment (ERGIC) also play critical roles in autophagy, a process tightly linked to lysosomal recovery. The ER and ERGIC are well-known sources of membranes for autophagosome biogenesis, supplying lipids and proteins required for phagophore expansion (Han et al., 2023; Melia et al., 2020). Building on these findings, the interplay between ER/ERGIC-driven autophagosome formation and lysosomal repair mechanisms—such as lysophagy—remains an open question. For instance, it is unclear whether ER-derived autophagosomes are preferentially recruited to engulf damaged lysosomes or if their maturation is synchronized with lysophagy. Elucidating these interactions could clarify how membrane trafficking pathways converge to restore lysosomal function, offering insights into the integration of autophagy and organelle repair. By exploring these mechanisms, researchers could refine models of lysosomal recovery, emphasizing the ER’s dual role in calcium regulation and membrane supply, and how these functions are coordinated to resolve lysosomal stress.
Golgi apparatus and lysosomal damage
In contrast to the ER and ERGIC, which directly interact with lysosomes through membrane contact sites to repair damage, the Golgi apparatus engages with lysosomes in a more indirect and unidirectional manner. This interaction primarily occurs via two pathways: 1) Protein Transport: Proteins, including hydrolases processed by the Golgi apparatus, are transported to endosomes through vesicular trafficking. These proteins ultimately reach lysosomes through acidification and fusion within the endolysosomal pathway (Melia et al., 2020; Scheuring et al., 2011). Therefore, deficiencies in posttranslational modifications within the Golgi apparatus are strongly associated with lysosomal dysfunction-related diseases (Akaaboune and Wang, 2024; Sou et al., 2024). 2) Autophagosome Biogenesis: The Golgi apparatus also serves as a source of key proteins and membrane components necessary for autophagosome formation (Gao et al., 2016; Sawa-Makarska et al., 2020). These components are incorporated into autophagosomes, which subsequently fuse with lysosomes to form autolysosomes, thereby delivering their cargo for degradation.
Despite these established pathways, the interactions between the Golgi apparatus and lysosomes might be more complex. In numerous diseases characterized by lysosomal dysfunction—such as lysosomal storage disorders (Lojewski et al., 2014; Shammas et al., 2019), neurodegenerative diseases (Gosavi et al., 2002; Martínez-Menárguez et al., 2019), COVID-19 infection (Cortese et al., 2020; Zhang et al., 2024), nicotine exposure (Govind et al., 2021), and epilepsy or other mental disorders triggered by electrical signal disturbances (Thayer et al., 2013)—morphological alterations in the Golgi apparatus have been consistently observed. These alterations include fragmentation (reduction in Golgi stack organization and increased dispersal), vesiculation (increased formation of Golgi-derived vesicles), and depolarization (randomized Golgi distribution), indicating that lysosomal damage and Golgi apparatus disorganization are concomitant events in these pathologies.
Similar to the ER, several Golgi-resident proteins may be recruited to or localized near damaged lysosomes, further suggesting a more complex interplay between the Golgi apparatus and lysosomal dysfunction. For example, Rab34, a Golgi-localized protein, plays a significant role in lysosome positioning and function. Overexpression or constitutive activation of Rab34 relocates lysosomes to the peri-Golgi area (Kumar et al., 2024; Wang and Hong, 2002) and facilitates the fusion of phagosomes with lysosomes (Seto et al., 2011). Additionally, loss-of-function variants of Rab34 are associated with various ciliopathies (Batkovskyte et al., 2024; Bruel et al., 2023), suggesting the possibility that dysregulation of Rab34-mediated Golgi-lysosome interactions may contribute to the pathogenesis of human diseases. The possibility of Golgi–lysosome interaction is further supported by single-organelle immunoprecipitation–coupled mass spectrometry studies of the Golgi (Fasimoye et al., 2023) and lysosomes (Eapen et al., 2021; Jia et al., 2022; Tan and Finkel, 2022; Wyant et al., 2018), underscoring the need for continued investigation.
Discussion
Lysosomal quality control is vital for cellular homeostasis and disease progression, with lysosomes employing alternative secretion pathways, such as exocytosis, exosome release, and secretory autophagy, when traditional repair mechanisms fail. Interactions with the endomembrane system, particularly the ER and Golgi apparatus, are also essential for lysosomal repair and function. These pathways play key roles in diseases, such as neurodegenerative disorders, where exosome-mediated spread of aggregated proteins accelerates progression, and cancer, where lysosomal exocytosis contributes to drug resistance and metastasis. Pathogens also exploit lysosomal secretion to enhance survival and spread, highlighting its complex role in disease.
Targeting lysosomal secretion and enhancing organelle interactions offer promising treatment strategies for lysosome-related diseases. Modulating exosome release could limit pathogenic protein spread in neurodegeneration, while inhibiting lysosomal exocytosis may help overcome cancer drug resistance. Strengthening ER-lysosome and Golgi-lysosome interactions could enhance lysosomal resilience in various diseases. These approaches aim to mitigate lysosomal dysfunction and improve cellular stress responses, offering new therapeutic perspectives.
Despite progress, key questions remain in lysosomal biology. Future research should focus on how lysosomal positioning impacts quality control, identifying specialized lysosome subpopulations, and understanding coordination with organelles like mitochondria. Additionally, the molecular triggers behind Golgi morphological changes in response to lysosomal damage and their role in repair need further investigation. Addressing these questions is critical to fully understanding lysosomal function and its integration within the cellular network.
In conclusion, lysosomes employ diverse strategies, including alternative secretion pathways and organelle interactions, to maintain cellular homeostasis under stress. These mechanisms are crucial in diseases like neurodegeneration, cancer, and infections. A deeper understanding of lysosomal-endomembrane interactions will uncover new therapeutic targets and help improve strategies for managing diseases linked to lysosomal dysfunction.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Abe T.Kuwahara Suenaga T.Sakurai S.Takatori M. S.Iwatsubo T. (2024). Lysosomal stress drives the release of pathogenic α-synuclein from macrophage lineage cells via the LRRK 2-Rab 10 pathway. i Science 27, 108893. 10.1016/j.isci.2024.108893 38313055 PMC 10835446 · doi ↗ · pubmed ↗
- 2Aits S.JäätteläM. (2013). Lysosomal cell death at a glance. J. Cell Sci. 126, 1905–1912. 10.1242/jcs.091181 23720375 · doi ↗ · pubmed ↗
- 3Akaaboune S. R.Wang Y. (2024). Golgi defect as a major contributor to lysosomal dysfunction. Front. Cell Dev. Biol. 12, 1386149. 10.3389/fcell.2024.1386149 38721528 PMC 11076776 · doi ↗ · pubmed ↗
- 4Almeida M. F.Bahr B. A.Kinsey S. T. (2020). Endosomal-lysosomal dysfunction in metabolic diseases and Alzheimer's disease. Int. Rev. Neurobiol. 154, 303–324. 10.1016/bs.irn.2020.02.012 32739009 PMC 8428780 · doi ↗ · pubmed ↗
- 5Alvarez-Erviti L.Seow Y.Schapira A. H.Gardiner C.Sargent I. L.Wood M. J. (2011). Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis. 42, 360–367. 10.1016/j.nbd.2011.01.029 21303699 PMC 3107939 · doi ↗ · pubmed ↗
- 6Bakowski M. A.Braun V.Lam G. Y.Yeung T.Heo W. D.Meyer T. (2010). The phosphoinositide phosphatase Sop B manipulates membrane surface charge and trafficking of the Salmonella-containing vacuole. Cell Host Microbe 7, 453–462. 10.1016/j.chom.2010.05.011 20542249 · doi ↗ · pubmed ↗
- 7Ballabio A.Bonifacino J. S. (2020). Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 21, 101–118. 10.1038/s 41580-019-0185-4 31768005 · doi ↗ · pubmed ↗
- 8Bao F.Zhou L.Zhou R.Huang Q.Chen J.Zeng S. (2022). Mitolysosome exocytosis, a mitophagy-independent mitochondrial quality control in flunarizine-induced parkinsonism-like symptoms. Sci. Adv. 8, eabk 2376. 10.1126/sciadv.abk 2376 35417232 PMC 9007515 · doi ↗ · pubmed ↗