Influence of apolipoprotein E genotype on the proteomic profile in cerebral microdialysis after human severe traumatic brain injury: a prospective observational study
Caroline Lindblad, Andrea Klang, David Bark, Cristina Bellotti, Anders Hånell, Per Enblad, Anders Lewén, Elham Rostami

TL;DR
This study explores how the apolipoprotein E genotype affects protein levels in the brain after severe traumatic brain injury, finding that protein levels are strongly influenced by time and genotype.
Contribution
The study is the first to examine the influence of apolipoprotein E genotype on microdialysate protein expression in clinical traumatic brain injury.
Findings
Protein levels in cerebral microdialysate are strongly dependent on time from trauma.
Two proteins showed levels dependent on both time and apolipoprotein E genotype.
Aβ42/40 ratio was not related to time or apolipoprotein E genotype.
Abstract
Patient-tailored treatment, also known as precision-medicine, has been emphasized as a prioritized area in traumatic brain injury research. In fact, pre-injury patient genetic factors alone account for almost 26% of outcome prediction variance following traumatic brain injury. Among implicated genetic variants single-nucleotide polymorphism in apolipoprotein E has been linked to worse prognosis following traumatic brain injury, but the underlying mechanism is still unknown. We hypothesized that apolipoprotein E genotype would affect the levels of pathophysiology-driving structural, or inflammatory, proteins in cerebral microdialysate following severe traumatic brain injury. We conducted a prospective observational study of patients with severe traumatic brain injury treated with invasive neuromonitoring including cerebral microdialysis at Uppsala University Hospital. All patients were…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
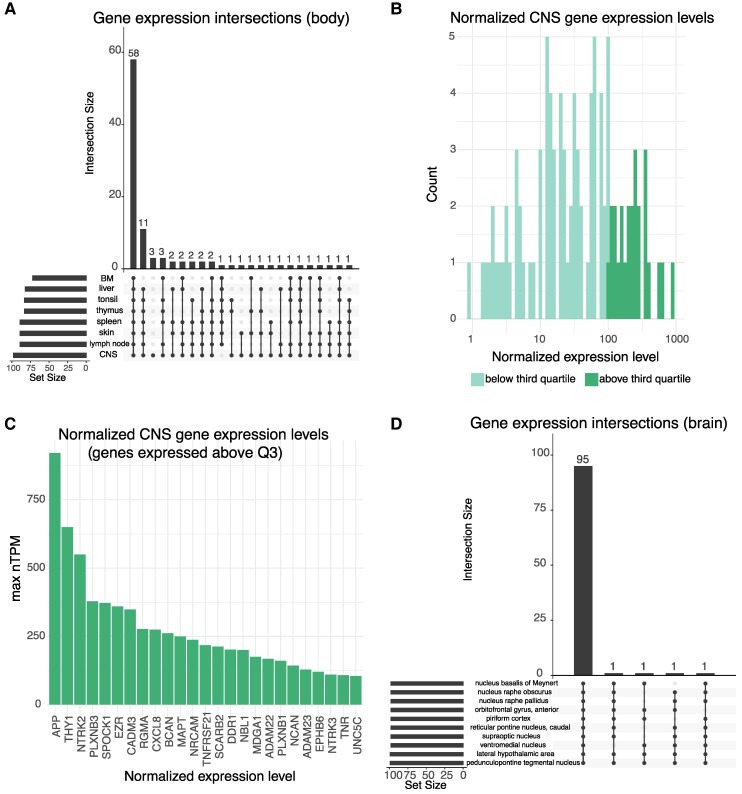 Figure 1
Figure 1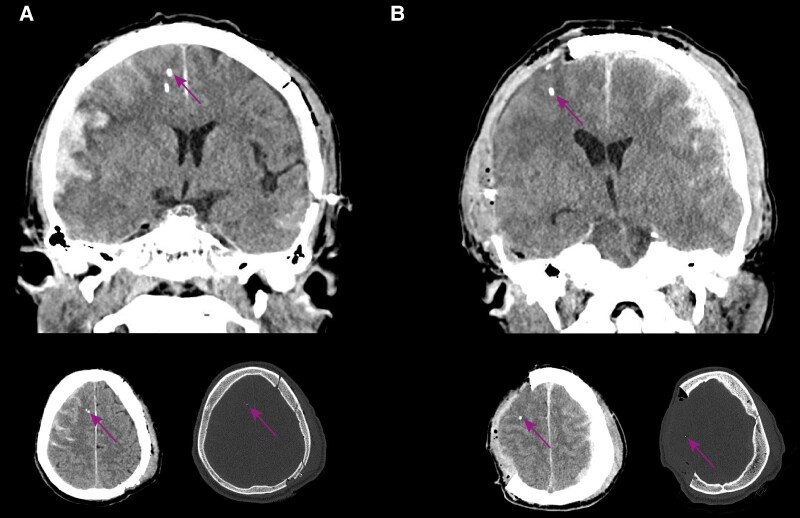 Figure 2
Figure 2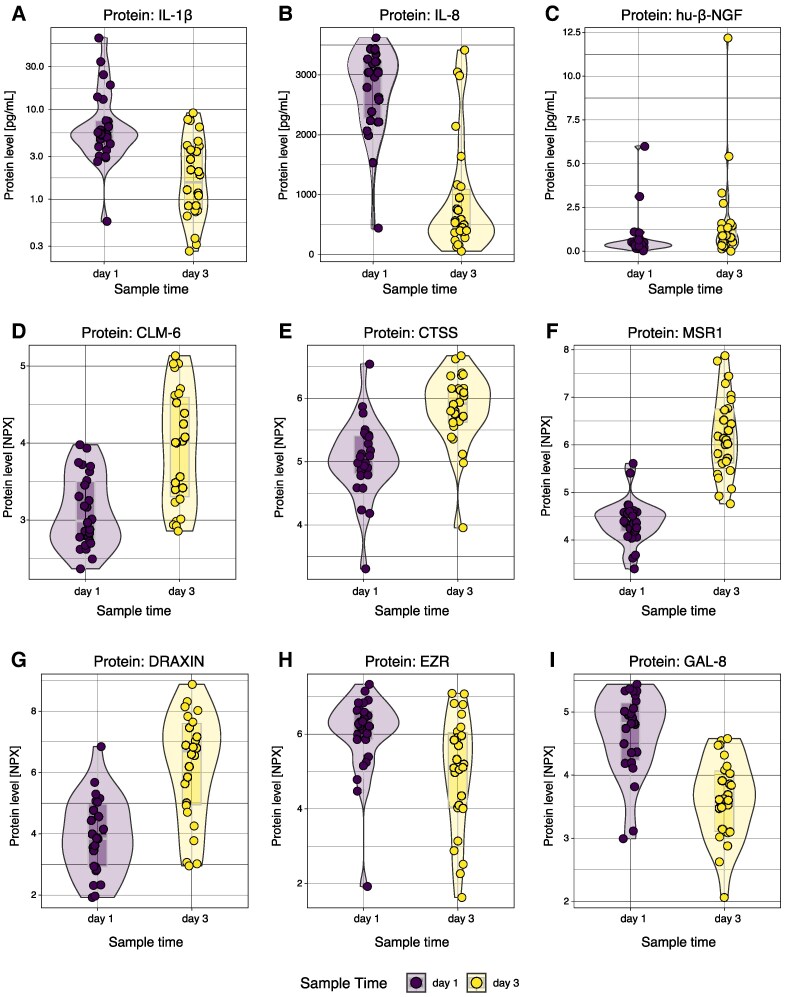 Figure 3
Figure 3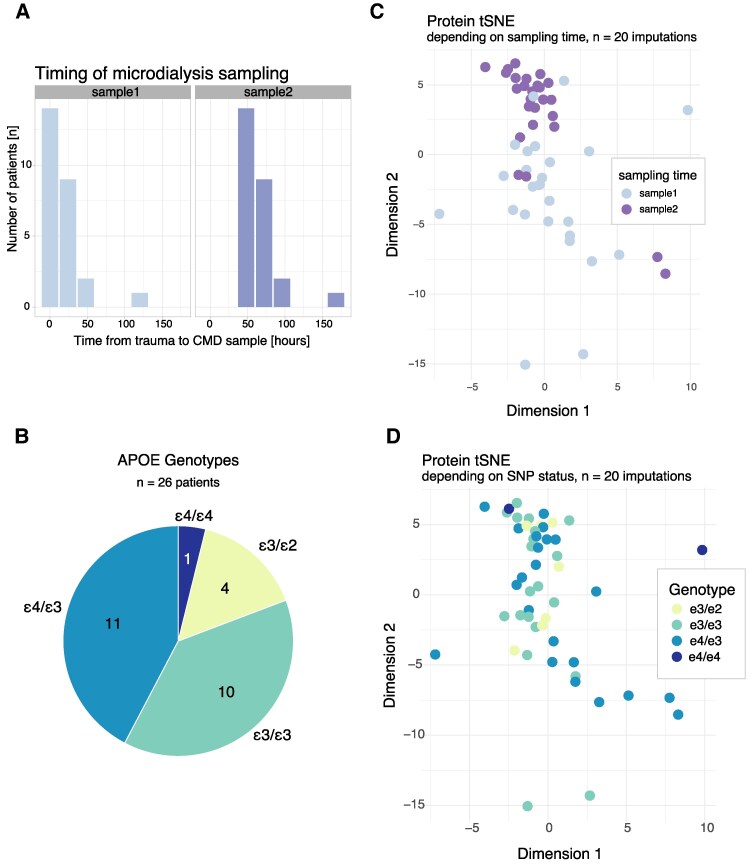 Figure 4
Figure 4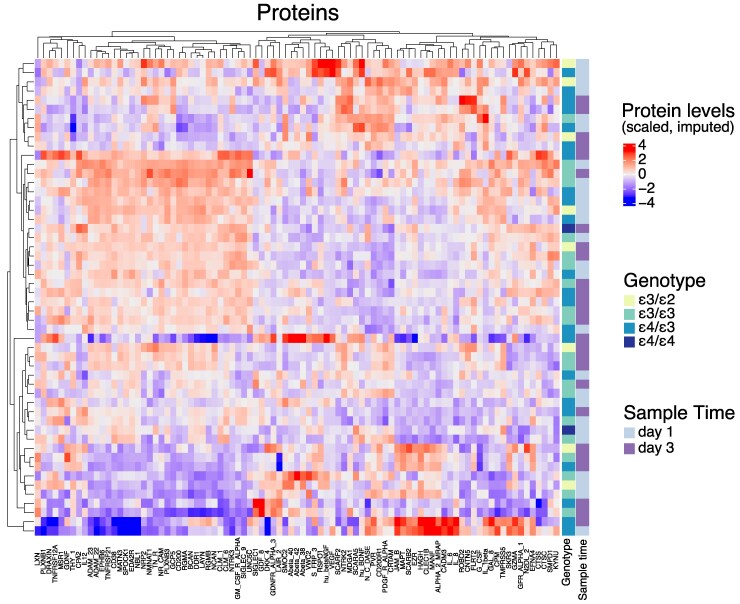 Figure 5
Figure 5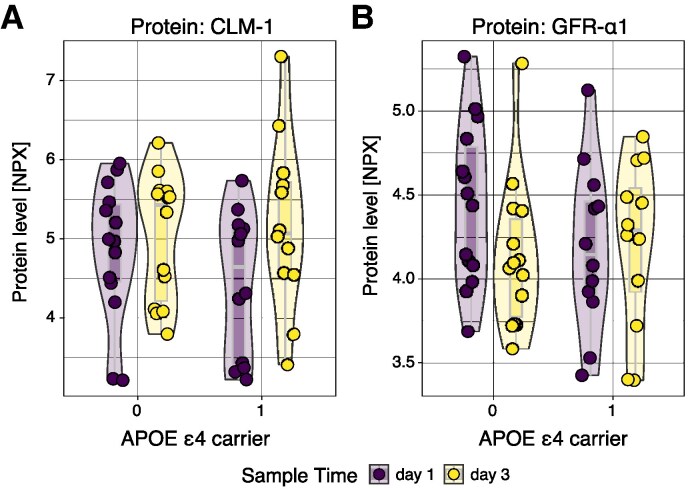 Figure 6
Figure 6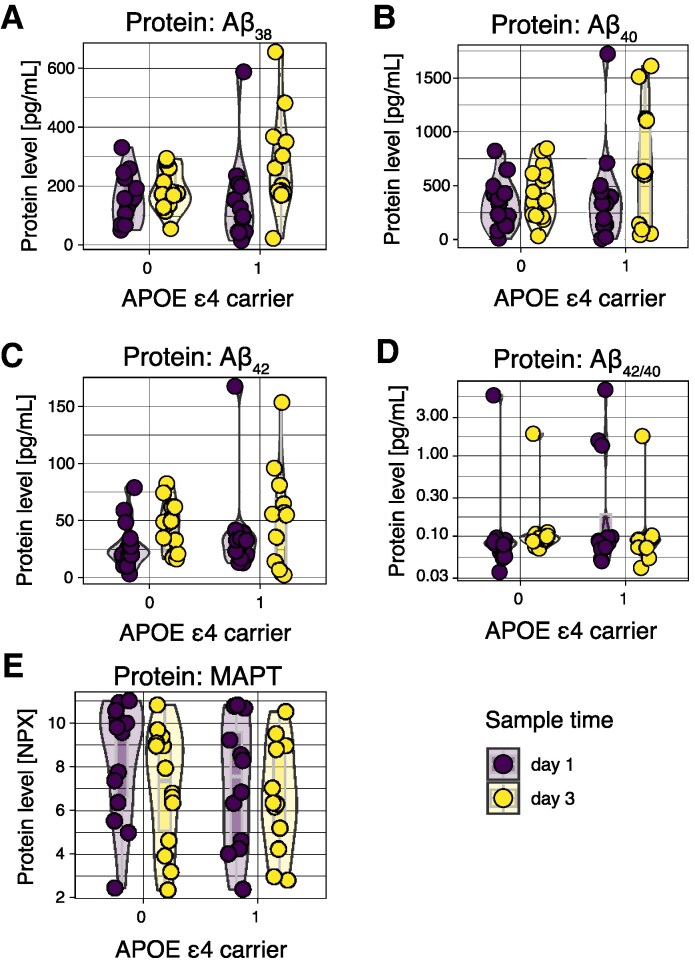 Figure 7
Figure 7| Variable | TBI cohort ( | Unit/metric |
|---|---|---|
| Age | 45 (18) | Years |
| Sex (Male) | 19 (73) | Count (%) |
| Pupils | Bilaterally normal: 22 (85) | Count (%) |
| Glasgow coma scale (admission) | 7 (7–9) | Arbitrary unit (median, interquartile range [IQR]) |
| Glasgow Coma Scale Motor Score (admission) | 5 (5–5) | Arbitrary unit (median, IQR) |
| Anticoagulant therapy | 2 (7.7) | Count (%) |
| Injury mechanism | Assault: 1 (3.8) | |
| Dominant injury pattern | Fracture of the calvarium: 1 (3.8) | Count (%) |
| Pentobarbital coma throughout NCCU stay | 2 (7.7) | Count (%) |
| Days in respirator | 13 (9–18) | Days (median, IQR) |
| Months to follow-up | 6 (5–7) | Months (median, IQR) |
| Glasgow Outcome Scale Extended | 1. Dead: 1 (3.8) | Count (%) |
| Glasgow Outcome Scale Extended (dichotomized) | Favourable: 12 (46) | Count (%) |
| Variable | TBI cohort ( | Unit/metric |
|---|---|---|
| Anatomical region | Frontal lobe: 24 (92) | Count (%) |
| Hemisphere | Right: 20 (77) | Count (%) |
| Distance from cortical surface | 18 (12) | Millimetre (mean, SD) |
| Distance from closest lesion | 36 (22) | Millimetre (mean, SD) |
| Distance from largest lesion | 42 (25) | Millimetre (mean, SD) |
| Type of largest lesion used for distance measurement | Extra-axial bleeding (non-evacuated): 4 (15) | Count (%) |
- —Uppsala County10.13039/501100009230
- —Uppsala University10.13039/501100007051
- —Swedish state
- —Knut and Alice Wallenberg Foundation10.13039/501100004063
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAcute Ischemic Stroke Management · Traumatic Brain Injury and Neurovascular Disturbances · Intracerebral and Subarachnoid Hemorrhage Research
Introduction
Precision-medicine has been emphasized as a prioritized area in traumatic brain injury (TBI) research.^1,2^ Among putative factors eligible for personalized treatment considerations, pre-injury patient characteristics are likely to be important. In fact, patient genetic set-up alone accounted for almost 26% of outcome prediction variance utilizing Glasgow Outcome Score Extended (GOSE)^3^ 6 months post-TBI.^4^ Several diverging genetic variants have been implicated,^5^ but genetic variations in the apolipoprotein E (APOE) stands out as particularly well-studied.^6^ In the CNS, APOE is a predominantly astrocytic protein encoded by the APOE gene located at chromosome 19^7,8^ with two commonly occurring single-nucleotide polymorphisms (SNPs) at position 112 and 158 causing the replacement of cysteine with arginine.^8^ In total, there are three different APOE alleles—APOE ɛ2, ɛ3 and ɛ4, thus enabling six different genotypes.^8,9^ In the absence of head injury, the ɛ4 allele has been attributed to exhibit a dose-dependent increased risk for Alzheimer's disease.^10^ Interestingly, in small TBI cohorts of varying injury severity, APOE ɛ4 carriers have been observed to have a worse prognosis.^11,12^ These findings have later been corroborated in meta-analyses with robustly incremented patient cohorts.^6,13^
The exact mechanism by which APOE ɛ4 is deleterious is still unknown. Recently, APOE ɛ4 carriers without head injury were shown to exhibit blood–brain barrier (BBB) breakdown and activation of the matrix metalloproteinase 9 pathway.^14^ APOE ɛ4 has also been associated with heightened microglial activation and other neuroinflammatory mediators^15^ in a cohort of non-TBI subjects.^15,16^ We have previously investigated BBB disruption, and neuroinflammatory pathways including matrix metalloproteinase 9 in CSF following severe TBI,^17^ but without finding any relationship to APOE ɛ4. One opportunity onwards to understand APOE-associated pathophysiology is to use cerebral microdialysis,^18^ utilized to focally monitor metabolism in the cerebral extracellular space,^19^ while it also allows for studies of e.g. neuroinflammation and directed proteomic investigations.^20^ With this in mind, we hypothesized that APOE ɛ4 allele homozygosity or heterozygosity would influence focal levels of proteins in brain extracellular fluid (i.e. microdialysate) following severe human TBI. We did not assume that protein levels would be related to APOE ɛ4 in a dose-dependent manner. We, therefore, aimed to undertake antibody-based proteomic profiling of microdialysate in a human severe TBI cohort, where the APOE genotype was known.
Materials and methods
This was a prospective, observational study undertaken at the neurocritical care unit (NCCU) at Uppsala University Hospital (Uppsala, Sweden) between December 2014 and September 2020. Written informed consent was obtained from next-of-kin. The study was undertaken in accordance with the Declaration of Helsinki and Swedish law. Ethical approval was granted by the Uppsala County branch of the Swedish Ethical Review Authority (#2010/138, #2010/138/1, #2010/379, #2015/224 and #2020-05462).
Patient inclusion, management and sample size considerations
All patients included were 15 years or older and had sustained a severe TBI, which necessitated multi-modal invasive intracranial monitoring including cerebral microdialysis. A TBI was defined as ‘an alteration in brain function, or other evidence of brain pathology, caused by an external force’.^21^ A severe TBI was defined clinically as a Glasgow Coma Scale (GCS) ≤ 8,^22,23^ which although imperfect is congruent with recent international multi-centre collaborative efforts.^2^ In addition, patients with a GCS > 8 but with neuroradiological signs on computerized tomography evocative of impending deterioration or increased intracranial pressure such as compressed basal cisterns, mid-line shift >5 mm and/or an un-evacuated mass lesion, or smaller lesions concurrently co-existing^24^ were also deemed as a severe TBI. These findings were routinely assessed by the neurosurgeon together with the neuroradiologist on-call in our centre. These radiological findings are also well in-line with formal CT classification systems,^25^ of which the original Marshall classification system^26^ was specifically developed to find patients with a mis-match between their clinical status and neuroradiological imagery. Sample size estimation was decided utilizing currently available literature, where several studies have investigated neuroinflammatory proteins in brain extracellular fluid from severe TBI patients.^27-29^ Two studies were observational^28,29^ and included n = 10 patients, whereas one^27^ was a follow-up study to a phase II randomized controlled trial^30^ that included n = 20 patients in total. In total, we therefore sought to recruit >20 patients. Patient enrolment was dependent on senior author presence, thus leading to non-consecutive enrolment of subjects into the study. Data on patients not enrolled are not available.
Uppsala University Hospital has a catchment area of 1.5–2 million citizens. Patients are either admitted directly from our hospital or transferred from regional hospitals across the catchment area, where initial patient stabilization and resuscitation are undertaken before transfer to our department. Patient management is undertaken in accordance with the Brain Trauma Foundation Guidelines,^31^ meaning that intracranial pressure (ICP) is maintained <20 mmHg, while cerebral perfusion pressure is maintained at >60 mmHg. In addition, a standardized clinical management protocol focused on secondary insult prevention has been implemented and shown to improve long-term outcome.^32^ In brief, patients were mechanically ventilated and received propofol and morphine for sedation and analgesia. We aimed for PaO_2_ ≥ 12 kPa. Patients with unstable ICP were initially hyperventilated, aiming at a PaCO_2_ range of 4.0–4.5 kPa. Patients were gradually normo-ventilated as soon as ICP permitted. For all patients, systolic blood pressure was targeted to be >100 mmHg. All patients obtained neuromonitoring equipment comprising an intracranial pressure monitor entailing either an external ventricular drain (HanniSet, Xtrans, Smith Medical GmbH), or an intra-parenchymatous monitor (Codman ICP Micro-Sensor, Codman & Shurtleff). The cerebral microdialysis catheter was inserted concurrently with the ICP device via the same burr-hole or craniotomy as the ICP device. The implantation site of choice was the non-dominant (usually right) frontal lobe, ca. 1–2 cm anteriorly to the coronal suture or else the same as the location of the craniotomy. A needle-based corticotomy was done, and the catheter was inserted at ∼20–30 mm depth. A 71 high cut-off brain microdialysis catheter with a membrane length of 10 mm and a membrane cut-off of 100 kDa (M Dialysis AB, Stockholm, Sweden) was used. We utilized perfusion fluid CNS (M Dialysis AB) containing NaCl (147 mmol/L), KCl (2.7 mmol/L, CaCl_2_ (1.2 mmol/L) and MgCl_2_ (0.85 mmol/L). Across the study period, we initiated perfusion fluid supplementation with human serum albumin^33^ followed by 3% dextran, where the latter has been shown to improve inflammatory marker recovery in vitro.^34^ A standard perfusion rate of 0.3 μL/min was achieved using a 106 Microdialysis pump (M Dialysis AB) and analysed utilizing either the ISCUSflex microdialysis analyzer or the CMA 600 analyzer (M Dialysis AB). As a routine, the microdialysis monitoring should continue for at least 5 days. When the patient was stable and the neurointensive care was completed, the patient was either moved to a step-down unit or discharged to the referring hospital. Following discharge, patients were followed-up at 6–12 months through either a structured questionnaire or a telephone interview documenting the GOSE.^3^
Sample collection, data acquisition and raw-data processing
Patients were sampled from whole-blood via an arterial line and from brain extracellular fluid (microdialysate) upon the first and third day following trauma. DNA was extracted from 200 μL whole-blood by using the Qiagen QIAamp (Qiagen) blood mini kit. DNA quantification was performed utilizing the Invitrogen Qubit 1X dsDNA HS Assay kit (Thermo Fisher Scientific) and the DNA quality was assessed through the Agilent Tapestation genomic DNA screen tape (Agilent). All samples were of high quality (DNA Integrity Number > 7). PCR amplification (background read and allelic discrimination) was run with Mastermix TaqPath ProAmp (Thermo Scientific). PCR reactions were run in triplicates in 384-well plates in 5 μL reactions with 5 ng of DNA. Amplification, background read and allelic discrimination were performed on Applied Biosystem HT 7900 PCR-machine (Thermo Fisher Scientific). For SNP discrimination, the human TaqMan SNP Genotyping Assay (Thermo Fisher Scientific) with the catalogue numbers C_3084793_20 (rs429358) and C_904973_10 (rs7412) were used. Raw-data processing was undertaken via Thermo Fisher Scientific's analytical platform Thermo Fisher Connect™. APOE allele status was determined by combining the output of the two assays so that APOE ɛ2 allele denotes rs7412T/rs429358T (amino acids: cysteine 112/cysteine 158); APOE ɛ3 allele denotes rs7412 C/rs429358T (amino acids cysteine 112/arginine 158) and APOE ɛ4 allele denotes rs7412 C/rs429358 C (amino acids arginine 112/arginine158).^8,9^ All genotyping steps were undertaken via the Karolinska Institutet Bioinformatics and Expression Analysis Core Facility.
Microdialysis samples were analysed through a proximity extension assay (Olink, Uppsala, Sweden), quantifying n = 92 proteins implicated in the CNS or its disorders. All Olink assays were undertaken by the company. For one protein [Interleukin (IL)-12], the protein was matched with two different UniProt IDs,^35^ as the protein IL-12 occurs both as IL-12α and IL-12β. All protein levels are reported as ‘normalized protein expression’, an arbitrary unit on log_2_ scale derived from the company. Quality control in conjunction with the analysis was conducted per assay and sample through n = 4 internal controls. To fulfil quality control criteria, the sample concentration of the internal control should deviate ≤0.3 from the median control concentration. All samples passed quality control, but several samples exhibited specific protein levels below the limit of detection. For each instance where the protein concentration was below the lower limit of detection, the protein concentration value was encoded as a missing value.
In addition, n = 9 proteins not available for proximity extension assay were quantified in microdialysate through multi-array technology, i.e. a combination between a protein, antibody-based array and electrochemiluminescence via Meso Scale Discovery Inc. (Maryland, USA), who also undertook all sample processing. We utilized the pre-designed V-plex Ab peptide panel for the neurodegenerative proteins Amyloid-β (Aβ)-38, -40 and -42 (Meso Scale Discovery Inc.). Aβ-38, -40 and -42 are all fragments from the Aβ precursor protein and are thus reported together for tissue enrichment descriptions. We also customized a U-plex panel for quantifications of brain-derived neurotrophic factor and human nerve growth factor (β-NGF); as well as the neuroinflammatory proteins IL-1β, -6, -8 and vascular endothelial growth factor (VEGF) (Meso Scale Discovery Inc.). For meso scale analyses, all protein concentrations (reported in picograms per millilitre) were derived through backfit calculation using a standard curve. Values below or above the fitted curve range were considered as missing values, while values within the curve fit but outside of the optimal detection range were included.
A comprehensive overview on the analysed proteins, including Uniprot and Ensembl ID is found in Supplementary Table 1. Normal tissue expression and regional relative tissue enrichment have been robustly described through the Human Protein Atlas (HPA) effort.^36-39^ We used two public RNA sequencing datasets from the HPA (version 23) with Ensembl annotations (version 109), available through https://v23.proteinatlas.org/about/download (files: rna_tissue_consensus.tsv; rna_brain_hpa.tsv) to determine relative tissue enrichment (reported as the normalized tissue expression levels)^40^ of the proteins that we measured (Fig. 1A). Utilizing this, we set a cut-off of 0.1^40^ to exclude non-enriched proteins. Notably, several of the proteins assessed were enriched across both the CNS and the immune system under homeostatic conditions. Among the protein-encoding genes that we studied, we report HPA-derived CNS expression levels (Fig. 1B), as well as the upper quartile of CNS-enriched genes (Fig. 1C), and their regional CNS expression under homeostasis (Fig. 1D).
Baseline characteristics of the investigated proteins. In total, n = 101 proteins were investigated. At baseline, the genes coding for these proteins have a distinct enrichment in both the CNS and lymphoid tissues across the body (A). Among CNS-enriched genes in our data (B), n = 25 had the 25% highest expression levels (C). These proteins are, under homeostatic conditions expressed at the gene level in a multitude of CNS sub-regions (D). BM, bone marrow; CNS, central nervous system. Full protein names can be found in Supplementary Table 1.
Statistical analysis
All statistical operations were conducted utilizing R version 4.3.1,^41^ through the interface Rstudio version 2023.09.0 + 463. Continuous variables are presented as mean (SD) if normally distributed or else median (inter-quartile range). Categorical variables are presented as count (percentage). Unless stated otherwise, a P value ≤ 0.05 was considered significant. Across all operations, the tidyverse package^42^ was used. In addition, for data curation the janitor package^43^ was used. For graphical illustrations, the additional packages UpSetR,^44-46^ ggforce,^47^ RColorBrewer,^48^ cowplot,^49^ CompexHeatmap^50,51^ and circlize^52^ were used. Other relevant packages are cited in adjunct to specific operations where they were applied.
Missing data, most notably for the various proteins measured, were visualized graphically and computationally utilizing the visdat^53^ and naniar packages^54^ in addition to base R. In total, n = 12 proteins had missing values (for either of the two sampling time points) exceeding 70% of the total number of observations (Supplementary Table 2). This pertained to the proteins: NGF-β (measured through the proximity extension assay platform), CLEC10A, BMP4, NEP, WFIKKN1, FCRL2, IL-5R-α, LAT, CHD3, CDH6, NAAA and PRTG (full protein names described in Supplementary Table 1). These proteins were deemed too uncertain to impute and were therefore excluded from downstream analysis. For the remaining proteins, we imputed data utilizing the mice package.^55^ We undertook this analysis utilizing the pipeline suggested by one of the mice package creators.^56,57^ Hence, we imputed n = 20 datasets with n = 10 iterations. The method defaulted to by the mice package was predictive mean matching for the proteins. Imputations were conducted utilizing age and sex data on the study subjects as well as protein data. For downstream analyses, calculations and operations were conducted for each imputed dataset whereafter results were pooled.
Dimensionality reduction analysis was performed using t-distributed stochastic neighbouring (tSNE) algorithms, through the package Rtsne.^58^ Clustering analyses were undertaken utilizing the packages cluster,^59^ factoextra,^60^ NbClust,^61^ clValid^62^ and mclust.^63^ Clustering tendency of the data was evaluated utilizing the Hopkins statistic. The number of clusters and the clustering technique was determined by comparing n = 21 various clustering algorithms and defining the optimal number of clusters as the most common output from the different clustering algorithms. Clustering methods and number of clusters were evaluated utilizing three different clustering scores (Connectivity, Dunn and Silhouette).
For inferential analyses, we employed linear mixed models through the nlme package.^64^ Primarily, we were interested in protein levels as the dependent variable and APOE genotype (dichotomized into ɛ4 carrier or not) as the independent predictor variable, utilizing time from trauma and patient age as tentative covariates. We utilized uni- and multi-variable regression modelling. For multi-variable models, we employed a top–down modelling strategy, including all tentative predictors from univariate analysis including interaction terms, after which non-significant variables were sequentially removed so that the final model retained only significant variables. Across all models, we allowed random intercept but abstained from random slopes models, as we only had n = 2 time points for observations. We also abstained from defining an autocorrelation structure, as we utilized time as covariate. We also abstained from including distance from the microdialysis probe to the lesion as covariate, primarily because of the small sample size and risk for data overfitting. Analysis results are presented both in their unadjusted form (denoted punadjusted), and following Benjamini–Hochberg (i.e. the false discovery rate) multiple testing correction (denoted padjusted). Regression assumptions, such as residual variance homogeneity and residual normality, were modelled graphically. In addition, since we did not use any autocorrelation structure in the data, we also modelled the partial autocorrelation of model residuals, the latter through the forecast package.^65^ If the regression criteria seemed to be fulfilled graphically for the majority of proteins across the majority of imputations, the regression model was accepted to enable comparison between proteins.
Results
Patient and neuromonitoring characterization
In total, 26 patients were included and underwent invasive neuromonitoring as well as APOE genotyping (Table 1). Most patients were middle-aged men, who were unconscious at admission with a mixed injury pattern. Mortality in this cohort was notably lower than compared with reference literature.^66^ In total, 11 patients (42%, including subjects lost to follow-up) had an unfavourable long-term outcome at 6 (5–7) months follow-up.
Characteristics of the implanted cerebral microdialysis are shown in Fig. 2 and Table 2. Most commonly, the microdialysis probe was positioned in the non-dominant (usually right) frontal lobe (Table 2 and Fig. 2A). If the patient underwent a craniotomy in the same séance as the neuromonitoring equipment was implanted, the microdialysis was occasionally pragmatically put in the vicinity of the craniotomy (Fig. 2B).
Representative examples of localization of cerebral microdialysis. The cerebral microdialysis probe has a gold-tip coating,20 making it discernible on computerized tomography imaging. In A, a conventional non-dominant right frontal lobe placement of the probe is seen whereas in B, probe placement has been placed in adjunct to a hemicraniectomy. The arrows highlight the microdialysis tip. The other hyperattenuating tip is the parenchymal intracranial pressure monitor.
The microdialysis probe was implanted 18 (±12) mm from the cortical surface, and notably 36 (±22) mm from the closest injury lesion and 42 (±25) mm from the largest detectable lesion.
Protein levels in brain extracellular fluid follow a temporally distinct pattern and are dependent on patient age
We quantified 89 proteins and 1 protein ratio (Aβ42/40) longitudinally in cerebral microdialysate following TBI. Several proteins displayed a time-dependent trajectory upon univariate analysis without consideration of APOE status (n = 41, with punadjusted ≤ 0.05; n = 34 with padjusted ≤ 0.05; Supplementary Table 3). Normalized longitudinal trajectories are depicted for all significant proteins in Supplementary Fig. 1, while absolute protein concentrations for nine significant proteins are depicted in Fig. 3. Notably, as no control subjects were included in the study, the impact of the TBI on the Day 1 protein level is difficult to assess. The neuroinflammatory proteins IL-1β and IL-8 were seen to express higher values at Day 1 than at Day 3 following trauma (Fig. 3A and B). In accordance, it is well-known that innate immune proteins such as IL-β (Fig. 3A) increase early following trauma,^68^ reflecting microglia-mediated caspase-driven cleavage of pro-IL-1β into its mature form.^69^ IL-8 (Fig. 3B) has also been shown to be incremented closely following clinical TBI.^70^ Interestingly, both IL-1β and IL-8 have been linked to the production of β-NGF (Fig. 3C),^70,71^ and it has been suggested that astrocytes drive NGF production.^72^ Here, β-NGF demonstrated higher values at Day 3 than at Day 1, possibly speaking in favour of a delayed production. Other proteins, normally enriched in lymphoid regions of the body (CLM-6, CTSS and MSR1)^39,73^ (Fig. 3D–F), demonstrated higher values at Day 3 compared with Day 1 following trauma. CNS-enriched proteins^39,73^ (DRAXIN and EZR; Fig. 3G-H) demonstrated opposite trends. While DRAXIN (Fig. 3G) demonstrated higher values at Day 3 compared with Day 1 following trauma, EZR (Fig. 3H) showed the opposite trend. Other proteins, not necessarily primarily enriched in the immune system or CNS^39,73^ such as GAL-8 (Fig. 3I) also demonstrated time-dependent protein concentration alterations. Among all proteins investigated, n = 4 were dependent on patient age in univariate analysis, and comprised the proteins EDA2R, HAGH, MANF and SPOCK1 (Supplementary Table 4). None of these age-dependent proteins had protein levels dependent on time from trauma.
Protein expression in brain extracellular fluid is dynamic following severe TBI. Protein analysis was undertaken at the first and third day following trauma. Notably, several proteins of varying origin and functions display a longitudinal trajectory. In total, n = 41 proteins demonstrated a temporal trajectory (n = 34 proteins retained significance following multiple testing correction), of which a representative subset is demonstrated above (A-I). Here, one dot represents one study subject at one time point. Calculations were done utilizing a univariate linear mixed model with time as independent variable and patient identity as the random effect. Numerical results (t-statistics and P-values) are available in Supplementary Table 3. Here, no adjustment to APOE status is undertaken. All protein concentrations (except for IL-1β, IL-8 and β-NGF, where concentration is in picograms per millilitre) are provided in normalized protein expression (NPX) which is the arbitrary unit on the log2 scale utilized by the proximity extension assay company. Full protein names are displayed in Supplementary Table 1. d, day; NPX, normalized protein expression; TBI, traumatic brain injury.
Protein clustering depends on time from trauma, but not on APOE genotype
Cerebral microdialysis samples analysed for protein content at the first [0 (0–24) h] and third day [48 (48–72) h] following severe TBI are shown in Fig. 4A. All patients that underwent microdialysate sampling were genotyped for APOE SNP status (Fig. 4B). In large clinical datasets of patients, the ɛ3 allele is the most common, followed by firstly the ɛ4 and secondly the least common ɛ2 allele.^74^ This is partly mimicked in our data, where n = 14 patients (54%) express either ɛ3/ɛ3 or ɛ3/ɛ2. However, n = 12 patients (46%) expressed homozygosity or heterozygosity for ɛ4, which is probably higher than the general population. We next attempted clustering analysis utilizing tSNE to account for time and genotype (Fig. 4C and D). Whereas, patients’ protein content clearly clustered depending on time from trauma (Fig. 4C), there was no obvious clustering tendency depending on patient genotype (Fig. 4D).
tSNE analysis of cerebral microdialysate proteins. Cerebral microdialysate sampling was undertaken for n = 89 proteins at 1 day and 3 days following trauma (A) among patients with varying APOE genotypes (B). Cluster analysis utilizing tSNE was undertaken with regard to sampling time, where a clear clustering tendency was shown (C), as well as genotype, where however no obvious clustering tendency was shown (D). For C and D, the pooled results of all n = 20 imputations are shown. APOE, apolipoprotein E; SNP, single-nucleotide polymorphism; tSNE, t-distributed stochastic neighbour embedding.
To further explore this, we also depicted all protein levels for all patients depending on time and APOE status utilizing a heatmap (Fig. 5). Here, a weak clustering between proteins is discerned, but without any obvious relationship to patient genotype.
Heatmap of cerebral microdialysate proteins in relation to genotype. Cerebral microdialysis was undertaken on n = 89 proteins from n = 26 patients. Samples clustered with regard to protein content, but not obviously with relation to APOE genotype. d, day following trauma. Full protein names are displayed in Supplementary Table 1.
Two proteins have APOE SNP-dependent levels, following adjustment for time from trauma
We conducted regression analyses, modelling protein levels as dependent on the time elapsed since trauma and/or APOE ɛ4 carriership (homozygote or heterozygote). Upon interaction modelling, APOE ɛ4 carriership was a significant predictor of cerebral microdialysate levels of two proteins—the glial-derived neurotrophic factor (GDNF) family receptor α1 (GFRα1) and the protein CD300 molecule like family member f (CLM-1, also known as CD300f, CD300LF) (Fig. 6A-B). This was discerned as an interaction effect, for which the pooled P-values were P = 0.039 (CLM-1) and P = 0.020 (GFR-α1), respectively. CLM-1 levels were higher in non-APOE ɛ4 carriers at all time points, while GFR-α1 levels were only higher at the first day following trauma. For both, the interaction effect conferred increased protein levels over time, meaning that APOE ɛ4 carriership increased protein levels as time from trauma increased (Supplementary Table 5). Notably, CLM-1 is enriched in lymphoid organs^39,73^ and has been implicated as a protective factor in experimental (excitotoxic) brain injury.^75^ In contrast, GFR-α1 is the core receptor for GDNF, implicated in neurite outgrowth and branching.^76^ Similar results were found when conducting the same regression analysis but also adjusting for age. Notably, following multiple testing correction, no proteins retained significance.
Graphical output of interaction model of APOE ɛ4 carrier status and time for microdialysate protein level. Levels of both the immune protein CLM-1 (CD300LF and CD300f) (A), and the GDNF receptor GFR-α1 (B), are seen to be concomitantly dependent on time from trauma and APOE ɛ4 carrier status. CLM-1 (A) is higher among non-APOE ɛ4 carriers at both Day 1 and Day 3 following trauma, although there is a general increase in CLM-1 levels over time. Conversely, GFR-α1 levels (B) decrease over time among patients without APOE ɛ4, whereas protein levels increase longitudinally among APOE ɛ4 carriers. Here, one dot represents one study subject at one time point. Calculations were done utilizing a multi-variable mixed model with APOE ɛ4 carrier and time as independent variables presented as an interaction effect and patient identity as the random effect. Numerical results (β coefficients and P-values) are available in Supplementary Table 5. Full protein names are displayed in Supplementary Table 1. APOE, apolipoprotein; d, day; GDNF, glial-derived neurotrophic factor.
Inflammatory and neurodegenerative proteins exhibit time-dependent concentration levels following TBI, but do not seem to be related to APOE ɛ4 carrier status
APOE ɛ4 has been associated with inflammatory-mediated disease and BBB disruption as possible mechanisms prompting neurodegenerative disease.^10,14,15^ We assessed numerous inflammatory and BBB proteins, including the previously mentioned CLM-1, IL-1β and IL-8, but also IL-6 and VEGF. In contrast to IL-1β, APOE SNP status had a trend to predict IL-6 levels (punadjusted = 0.081). Upon multiple testing corrections, this effect was no longer significant. Protein levels of VEGF were not dependent on either time from trauma or APOE SNP status.
We also assessed the neurodegenerative proteins microtubule-associated protein tau (MAPT) and the amyloid-β (Aβ) peptides Aβ38, Aβ40, Aβ42 and the ratio Aβ42/40, as the latter has been evocative of intracerebral deposition of amyloid plaques (Fig. 7).^77^ The time elapsed since trauma predicted the levels of MAPT (punadjusted = 0.032, padjusted = 0.077; Supplementary Table 3) and Aβ40 peptide (punadjusted = 0.044, padjusted = NS; Supplementary Table 3). However, none of the other neurodegenerative protein markers, including the Aβ42/40 ratio, exhibited a significant association. APOE ɛ4 homozygosity or heterozygosity was not a predictor for microdialysate levels of these neurodegenerative proteins, neither upon univariate analysis nor adjusted for time upon multi-variate analysis.
Graphical output of regression model of APOE ɛ4 carrier status and time for neurodegenerative proteins assessed in microdialysate. Levels of Aβ38 (A) seem to be independent of time from trauma and APOE ɛ4 carrier status. Levels of Aβ40 (B) seem time-dependent (punadjusted = 0.044, padjusted = NS), while Aβ42 (C) and the ratio of Aβ42/40 (D) do not exhibit any strong trends. Protein levels of MAPT (E) were dependent on time (punadjusted = 0.040, padjusted = 0.096), but not genotype. Here, one dot represents one study subject at one time point. Calculations were done utilizing a univariate linear mixed model with time as independent variable and patient identity as the random effect. Numerical results (t-statistics and P values) are available for significant proteins in Supplementary Table 3. Full protein names are displayed in Supplementary Table 1. APOE, apolipoprotein; d, day; Aβ, amyloid-β; MAPT, microtubule-associated protein tau.
Discussion
This is the first report on brain extracellular fluid proteomics in relation to APOE SNP in 26 human patients with severe TBI. Brain extracellular fluid retrieved utilizing microdialysis demonstrated temporally distinct patterns of protein expression, reflecting severe TBI pathophysiology in the injured brain. We also characterized all proteins regarding SNP in APOE ɛ4 carriership, the currently known strongest genetic risk factor for late onset Alzheimer's disease^78^ and an independent predictor of poor outcome following severe TBI.^6,13^ Notably, acute (1 day and 3 days) protein levels of MAPT and Aβ peptides did not seem to be related to APOE genotype. In contrast, for the inflammatory protein CLM-1 and the neurotrophic protein GFR-α1, we found that protein levels in adjunct to time from trauma were dependent on carriership of APOE ɛ4, however, neither protein retained significance following multiple testing correction. This possibly reflects how a patient's genetic background also affects disease-processes following TBI. Our findings could be utilized for continuous fluid biomarker efforts, while they also entail novel pathophysiological data, laying the foundation for future disease-modifying studies as well as therapeutic implications, discussed in detail below.
Precision-based medicine warrants detailed pathophysiological portrayal in meticulously characterized clinical cohorts
Despite intense research efforts, treatment for severe TBI is directed to prevent secondary insult development rather than its underlying pathophysiology.^1,2^ In fact, some aspects of TBI pathophysiology are likely not actively monitored clinically. In line with this, the International Mission for Prognosis and Clinical Trial prognostic tool for assessment of functional neurologic outcome^79,80^ (via Glasgow Outcome Scale^81^) has an explained variance of ∼35%.^82^ This model contains exclusively pre-hospital and admission data, leading recent work to also include variables reflecting patient trajectories following admission.^83^ Even though this improves prediction to ∼52%, almost half of the variance in functional neurological outcome is still unaccounted for.^83^ We hypothesize that ongoing pathophysiology, such as neuroinflammation^69,84^ underlies at least some of this difference. The same holds true for pre-injury patient characteristics, such as specific genotypes, known to influence outcome.^4^
We therefore assessed n = 101 proteins in cerebral microdialysate following injury, of which n = 89 were eligible for analysis. We utilized two different platforms, one utilizing a proximity extension assay,^85,86^ and the other through a plate-based antibody-array coupled with electrochemiluminescence.^87^ Although it naturally would have been preferable to utilize one method, the current approach allowed us to profit from the highly multiplexed capability offered by proximity extension assays, while we could also assess proteins not yet reliably quantifiable utilizing this technique. This allowed us to examine a panel of structural neurological proteins, as well as inflammatory and neurodegenerative proteins. These proteins clustered with relation to time, and time was also an important predictor for the protein levels of 41 proteins in microdialysate. This mimics the sequential onset of neuroinflammation following TBI, known to occur in a strict time-dependent fashion.^88^ We can corroborate this in our data, by demonstrating the time-dependency of among else IL-1β and IL-8.^68,70^ Further, our results here also indicate that other structural (e.g. EZR and DRAXIN) as well as inflammatory proteins (e.g. CLM-6, CTSS and MSR1) follow a similarly stringent time-dependency. Jointly, this might indicate that the protein expression discerned is evocative of ongoing pathophysiology throughout the first 3 days following TBI. Importantly, our data shows that cerebral microdialysate detects these pathophysiological events. Onwards, this means that cerebral microdialysis might be utilized for monitoring of pathophysiology in the injured brain beyond the conventional detection of ischaemia. It would also be of interest to use microdialysis to monitor treatment response, as has been done both following metabolic and inflammatory modulation.^30,89^
APOE ɛ4 homozygosity or heterozygosity is related to focal protein expression, but does not influence MAPT or amyloid-β levels acutely
In adjunct to microdialysate protein quantifications, we characterized all patients with regard to APOE ɛ4 carrier status. Upon regression modelling, levels of two proteins (CLM-1 and GFR-α1) seemed to be influenced by an interaction effect between time and APOE status. In contrast to our findings, previous work utilizing CSF proteomics in a larger patient cohort did not show any convincing relationship between APOE ɛ4 status and protein levels.^17^ The discrepancy likely reflects that the underlying study design and protein panels were different. Yet, as our results did not retain significance following multiple testing corrections, they need to be replicated and then externally validated in an autonomous dataset.
We observed altered levels of the proteins CLM-1 and GFRα1. CLM-1 levels were higher in non-APOE ɛ4 carriers at all time points, while GFR-α1 levels were only higher at the first day following trauma. For both, the interaction effect conferred increased protein levels over time, meaning that APOE ɛ4 carriership increased protein levels as time from trauma increased. CLM-1 is an immune-system protein that in the CNS is expressed by glial cells.^75,90^ Recent data indicates that inhibition of CLM-1 is neurotoxic in vivo following an experimental penetrating TBI.^91^ In our data, APOE ɛ4 carriers seemed to have somewhat lower levels of CLM-1 both at Day-1 and -3 following trauma. In contrast to this, the GFRα1 is the core receptor for the neurotrophic peptide GDNF.^92^ Originally, GDNF-GFRα1 was found to support survival of mid-brain dopaminergic neurons and was thus of interest in Parkinson's disease research,^93^ but has since been seen to be important also for neuronal maintenance and plasticity in homeostasis.^94,95^ GDNF rather than GFRα1 has been investigated in experimental TBI^96,97^ and showed some promise. In our data, it would seem as if patients without the APOE ɛ4 allele express higher GFRα1 levels earlier, which speculatively could be beneficial for neuronal survival. Taken together, we show that protein levels might alter in relation to patient genotype. Speculatively, genotype-dependent levels of immune cells and neurotrophic cues could affect acute lesion development following TBI, thus possibly accounting for prognostic differences seen among patients with APOE ɛ4 SNP.
We also investigated the neurodegenerative proteins MAPT, Aβ38, Aβ40, Aβ42 and the ratio of Aβ42/40, the latter as it has been associated with amyloid plaque deposition in the brain.^77^ We did not see any relationship between either MAPT or amyloid-β peptide levels and APOE ɛ4 carrier status. SNP of APOE is the currently known strongest genetic risk factor for late-onset Alzheimer's disease,^78^ linked to BBB dysfunction.^14^ Numerous reports^6,7,11,13^ have described a link between APOE ɛ4 and poor outcome following TBI. Interestingly, APOE ɛ4 homozygosity or heterozygosity has also been linked to deficient repair of the BBB also following TBI.^98^ As BBB disruption following TBI seems to be influenced by neuroinflammatory pathways,^98^ and APOE is synthesized by astrocytes and microglia, two innate neuroinflammatory cells,^9^ it is reasonable to believe that neuroinflammation is altered because of APOE genotype. We therefore specifically quantified the levels of neuroinflammatory proteins following human TBI. Among neuroinflammatory proteins, IL-1β is a core cytokine mediator of the TBI inflammatory response.^69^ The IL-1 signalling pathway prompts downstream inflammatory activation, e.g. of IL-6.^99^ IL-6 has also been shown to be increased in CSF following human TBI,^17^ and is of relevance for cell interactions in vitro in neuroinflammatory model systems.^100^ We did not see any robust relationship between IL-1β or IL-6 and APOE SNP status. Taken together, the APOE genotype is observed to be related to certain acute protein alterations (CLM-1 and GFR-α1), but not to acute changes in conventional neuroinflammatory or neurodegenerative proteins.
Limitations
We acknowledge limitations with our study, most notably a small cohort size derived from a single centre. Study enrolment was non-consecutive, and we do not have access to data on patients not enrolled. Even though our cohort is similar to other TBI cohorts in the literature, we cannot exclude a systematic difference between patients enrolled and not-enrolled thus affecting external validity of our results. In addition, the limited cohort also likely yields the non-significant results upon multiple testing corrections, possibly reflecting a type II error. Yet, this cohort is amongst the most rigorously characterized in a severe TBI context, allowing hypothesis-generating work for future investigations in larger cohorts. Another limitation with our work is that cerebral microdialysis potentially is too focal a method to allow CNS-wide extrapolation,^20^ illustrated by earlier work that found limited overlap between protein quantifications in CSF, followed by cerebral microdialysis.^101^
Conclusion
Cerebral microdialysis reflects ongoing temporally distinct severe TBI pathophysiology. Whereas acute and subacute protein levels of Aβ peptides do not seem to be related to APOE genotype, both the neurotrophic receptor GFRα1 and the inflammatory receptor CLM-1 exhibited protein levels dependent on APOE genotype. This possibly reflects how a patient's genetic background also affects acute disease-processes following TBI, which might have therapeutic implications, but warrants further contextualization and external validation in larger clinical cohorts.
Supplementary Material
fcaf096_Supplementary_Data
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Maas AIR, Menon DK, Adelson PD, et al Traumatic brain injury: Integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 2017;16(12):987–1048.29122524 10.1016/S 1474-4422(17)30371-X · doi ↗ · pubmed ↗
- 2Maas AIR, Menon DK, Manley GT, et al Traumatic brain injury: Progress and challenges in prevention, clinical care, and research. Lancet Neurol. 2022;21(11):1004–1060.36183712 10.1016/S 1474-4422(22)00309-XPMC 10427240 · doi ↗ · pubmed ↗
- 3Jennett B, Snoek J, Bond MR, Brooks N. Disability after severe head injury: Observations on the use of the Glasgow Outcome Scale. J Neurol Neurosurg Psychiatry. 1981;44(4):285–293.6453957 10.1136/jnnp.44.4.285PMC 490949 · doi ↗ · pubmed ↗
- 4Kals M, Kunzmann K, Parodi L, et al A genome-wide association study of outcome from traumatic brain injury. E Bio Medicine. 2022;77:1–12.10.1016/j.ebiom.2022.103933 PMC 892784135301180 · doi ↗ · pubmed ↗
- 5Zeiler FA, Mc Fadyen C, Newcombe VFJ, et al Genetic influences on patient-oriented outcomes in traumatic brain injury: A living systematic review of non-apolipoprotein e single-nucleotide polymorphisms. J Neurotrauma. 2021;38(8):1107–1123.29799308 10.1089/neu.2017.5583 PMC 8054522 · doi ↗ · pubmed ↗
- 6Mc Fadyen CA, Zeiler FA, Newcombe V, et al Apolipoprotein E 4 polymorphism and outcomes from traumatic brain injury: A living systematic review and meta-analysis. J Neurotrauma. 2021;38(8):1124–1136.30848161 10.1089/neu.2018.6052 PMC 8054520 · doi ↗ · pubmed ↗
- 7Verghese PB, Castellano JM, Holtzman DM. Apolipoprotein E in Alzheimer’s disease and other neurological disorders. Lancet Neurol. 2011;10(3):241–252.21349439 10.1016/S 1474-4422(10)70325-2PMC 3132088 · doi ↗ · pubmed ↗
- 8Fernández-Calle R, Konings SC, Frontiñán-Rubio J, et al APOE in the bullseye of neurodegenerative diseases: Impact of the APOE genotype in Alzheimer’s disease pathology and brain diseases. Mol Neurodegener. 2022;17(1):62.36153580 10.1186/s 13024-022-00566-4PMC 9509584 · doi ↗ · pubmed ↗