A Novel Missense Variant of BMPR1A in Juvenile Polyposis Syndrome: Assessment of Structural and Functional Alternations
Mengyuan Yang, Ziyan Tong, Zhijun Yuan, Bingjing Jiang, Yingxin Zhao, Dong Xu, Ying Yuan

TL;DR
This paper reports a new BMPR1A gene variant linked to Juvenile Polyposis Syndrome, suggesting it affects protein stability and splicing, offering new insights into the disease's genetic basis.
Contribution
The study identifies a novel BMPR1A missense variant and proposes its impact on protein structure and function in Juvenile Polyposis Syndrome.
Findings
A novel BMPR1A missense variant (c.355C>T; p.R119C) was identified as pathogenic in Juvenile Polyposis Syndrome.
The variant is predicted to reduce molecular weight, affecting protein stability and posttranslational modifications.
The variant may also cause aberrant alternative splicing, contributing to disease mechanisms.
Abstract
Juvenile polyposis syndrome (JPS) is a rare precancerous condition associated with a high susceptibility to colorectal cancer. The genetic basis of JPS has been reported to lie in germline mutations in BMPR1A or SMAD4, resulting in diverse clinical manifestations and an elusive underlying mechanism. We firstly utilized a 139-gene next-generation sequencing (NGS) panel to detect the germline variants and further employed various prediction tools to assess the pathogenicity and functional alternations. Consequently, we identified a novel pathogenic BMPR1A missense variant (c.355C>T; p.R119C). More importantly, we proposed for the first time that the missense variant would lead to a decrease in molecular weight, potentially associated with reduced protein stability, diminished posttranslational modifications, and aberrant alternative splicing. These findings may provide novel perspectives…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
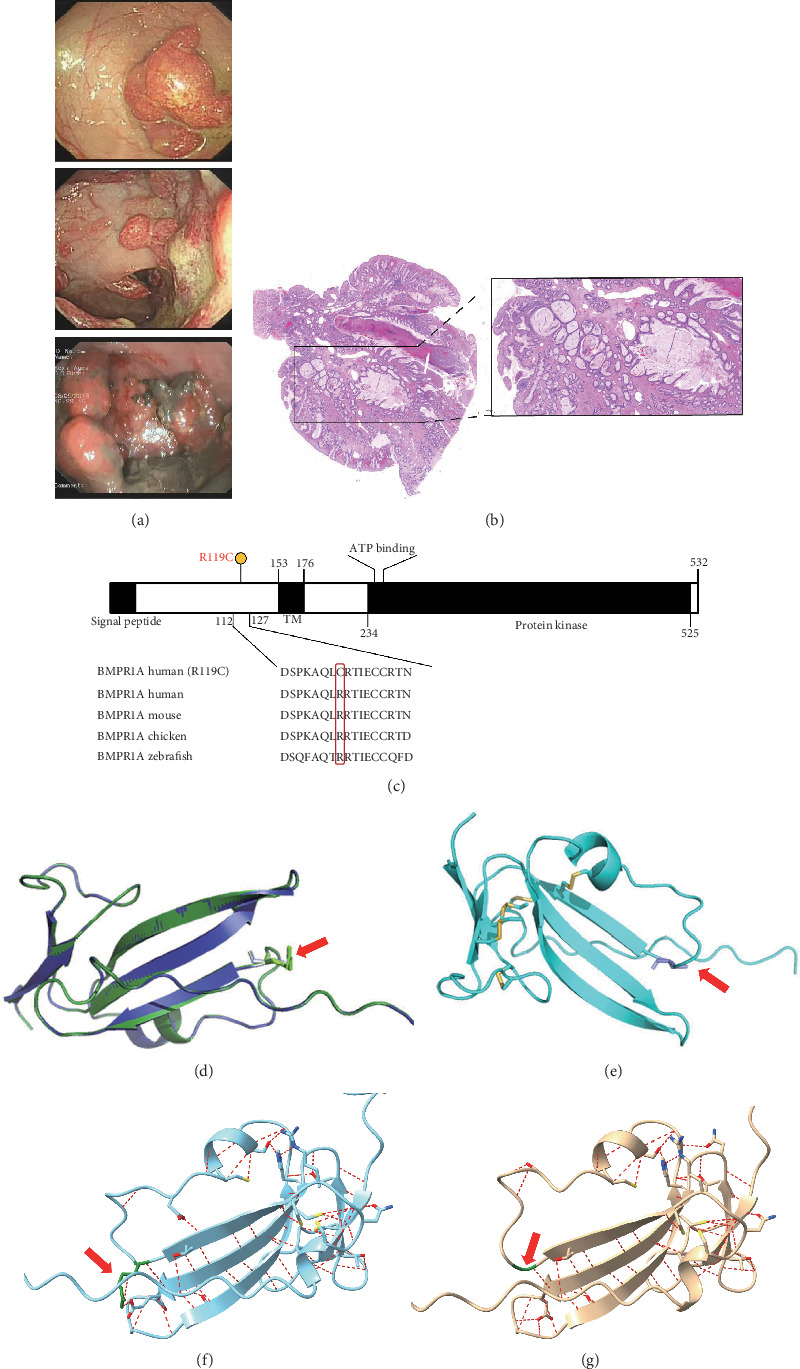 Figure 1
Figure 1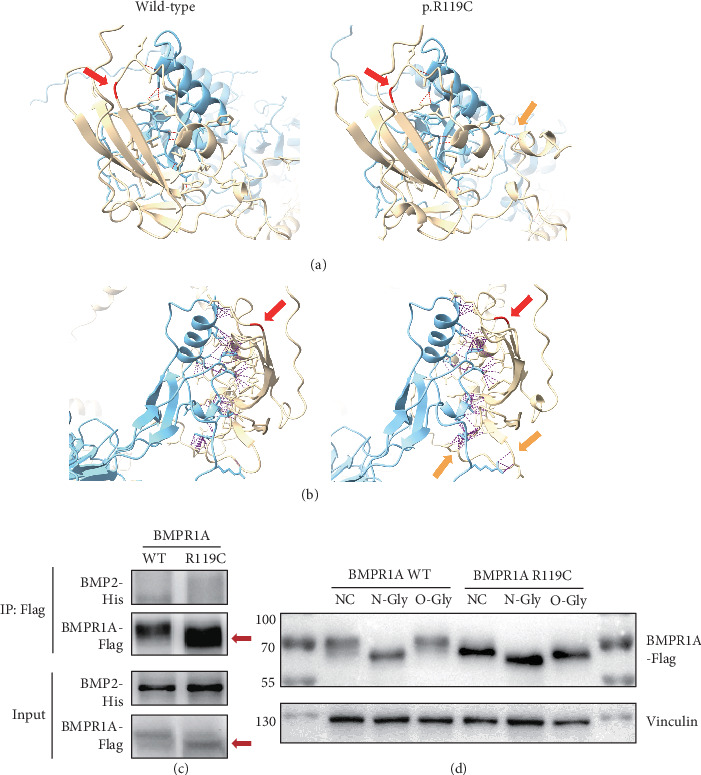 Figure 2
Figure 2- —Key Research and Development Program of Zhejiang Province
- —Beijing Xisike Clinical Oncology Research Foundation
- —Zhejiang Provincial Clinical Research Center for Cancer
- —National Natural Science Foundation of China
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic factors in colorectal cancer · Genomic variations and chromosomal abnormalities · Genomics and Rare Diseases
1. Introduction
Juvenile polyposis syndrome (JPS, OMIM Code 174900) is a rare autosomal dominant precancerous predisposition syndrome, with an incidence of approximately 1/100,000 [1, 2], typically resulting in multiple juvenile polyps in the colorectum (98%), stomach (14%), duodenum (7%), and small bowel (7%) [3]. These polyps significantly increase the risk of colorectal cancer, with a cumulative risk of up to 68% [4]. Germline disease-causing variants (DCVs) in the SMAD family member 4 (SMAD4) or bone morphogenetic protein receptor 1A (BMPR1A) genes have been reported in over 50% of JPS patients, with BMPR1A DCVs accounting for 17%–38% [5]. Despite the involvement of both proteins in the transforming growth factor-β (TGF-β)/bone morphogenetic protein (BMP) signaling pathway, these DCVs often result in distinct clinical presentations. JPS patients with SMAD4 mutations predominantly exhibit upper gastrointestinal polyps and hereditary hemorrhagic telangiectasia (HHT), while BMPR1A-mutated patients mainly present with colonic phenotypes [6]. It follows that the highly variable clinical phenotypes of JPS can be partially explained by these DCVs, including polyp locations, extraintestinal manifestations, and risk of malignant transformation [7]. However, the exact mechanism by which these DCVs influence the development of JPS has not been extensively investigated [1, 8, 9].
The BMPR1A protein, a serine–threonine kinase type I receptor of the TGF-β/BMP superfamily, regulates colonic epithelial differentiation through activation of SMAD4-mediated intracellular signaling [10]. Mutations in BMPR1A are associated with a variety of disorders including JPS, idiopathic pulmonary arterial hypertension [11], primary ovarian insufficiency [12], and abnormal skeletal development [13]. Currently, studies on the association between BMPR1A and JPS primarily focus on identifying novel DCVs and establishing the gene–phenotype correlation. In JPS, DCVs of BMPR1A encompass missense/nonsense variants, small insertions/deletions, and large genomic deletions [14, 15]. These variants tend to disrupt the TGF-β/BMP pathway and impact cell proliferation, playing a crucial role in pathogenesis [16]. Also, patients with contiguous deletions of phosphatase and tensin homolog (PTEN) and BMPR1A tend to develop JPS at an earlier age with more severity, indicating a synergistic effect of contiguous deletions [17, 18]. However, limited research has been conducted on specific pathogenic mechanisms. In Bmpr1a-mutant mice, polyps exhibited cystically dilated glands filled with mucin (a JPS-like phenotype), demonstrating the potential function of BMPR1A in a mouse model [19]. Furthermore, a functional study has suggested that certain BMPR1A missense mutations identified in JPS result in alterations to protein conformation and affect cellular localization [20]. Additionally, a novel nonsense variant (c.1114A>T, p.Lys372Ter) was found to cause nonsense-mediated mRNA decay (NMD), thus inhibiting BMP signaling activity [21]. More comprehensive functional experiments are necessary to validate the role of the BMPR1A-related signaling pathway in polyp formation and carcinogenesis of JPS.
Here, we present a case of JPS with BMPR1A missense variant (c.355C>T; p.R119C), proposing for the first time that this variant may lead to a decrease in molecular weight and can be interpreted as pathogenic.
2. Materials and Methods
2.1. Patients and DNA Samples
The study was approved by the Ethics Committee of the Second Affiliated Hospital of Zhejiang University School of Medicine (Approval No. 2017.066). Written informed consent was obtained from this patient.
Genomic DNA was extracted from peripheral EDTA-anticoagulated blood samples using the standard procedure (QIAamp DNA blood midi kit, Qiagen, Hilden, Germany).
2.2. Next-Generation Sequencing (NGS) and Analysis
The NGS panel covers the whole exon region of 139 genes associated with 16 cancers including colorectal cancer and 70 genetic syndromes, which was performed by Genetron Health (Beijing, China) on the HiSeqX-ten sequencing platform (Illumina, San Diego, United States). These 139 genes have been described in previous study [22]. We classified germline variants according to the American College of Medical Genetics and Genomics (ACMG) standards and guidelines for sequence variant interpretation [23].
2.3. Functional Experiments
2.3.1. Cell Culture
L293T cells were purchased from ATCC and were cultured at 37°C in 5% CO_2_ in the RPMI medium 1640 supplemented with 10% fetal bovine serum (FBS) (Gibco).
2.3.2. Construction of Overexpression Plasmid
The human BMPR1A (NM_004329) cDNA cloning was purchased from YouBio (Hunan, China), which was connected to the pcDNA3.1-3xflag according to the manufacturer's instructions. The missense variant c.355C>T was mutated through PCR amplification. The human BMP2 (NM_001200) cDNA cloning was purchased from YouBio (Hunan, China), which was tagged with 6xHis and then was connected to pcDNA3.1 according to the manufacturer's instructions.
2.3.3. Coimmunoprecipitation (CoIP)
The pcDNA3.1-BMP2-6xHis and pcDNA3.1-BMPR1A wildtype-3xflag or pcDNA3.1-BMPR1A mutant-3xflag plasmids were transfected into L293T cells at a 1:1 ratio. Transient transfection was performed by LipoD293 In Vitro DNA Transfection Reagent (SignaGen Laboratory, Rockville, Maryland, United States). Forty-eight hours after transfection, whole-cell extracts were lysed in IP buffer with protease inhibitor and incubated on ice for 30 min. After centrifugation for 15 min at 14,000 g, supernatants were collected and incubated with ANTI-FLAG M2 magnetic beads (M8823, Sigma-Aldrich, Darmstadt, Germany). After incubation overnight, beads were washed three times with immunoprecipitation buffer. After discarding the supernatant, add 1× loading buffer, and heat at 100°C for 5 min; the supernatant was magnetically separated for subsequent WB assay and blotted with mouse anti-FLAG (M2, F3165, Sigma-Aldrich, 1:1000) and mouse anti-6xHis (HIS.H8, Invitrogen, 1:1000).
2.3.4. Glycosylation Detection
The BMPR1A wildtype and mutant plasmids were transfected into L293T cells, respectively. Transient transfection was performed by LipoD293 In Vitro DNA Transfection Reagent (SignaGen Laboratory, Rockville, Maryland, United States). Forty-eight hours after transfection, cell proteins were extracted in 1× denaturing buffer. Prior to immunoblotting, samples were treated with N-glycosidase (N-Gly) F (PNGase F, P0704S) to remove N-glycans or O-glycosidase (O-Gly) (P0733S) and α2-3,6,8 neuraminidase (P0720S) to remove O-glycans, and these enzymes are all purchased from New England Biolabs (Beverly, Massachusetts). And then, the samples were blotted with mouse anti-FLAG (M2, F3165, Sigma-Aldrich, 1:1000) and rabbit anti-Vinculin (ET1705-94, Huabio, 1:1000).
2.4. Assessment of Functional and Structural Consequences
2.4.1. Prediction of Pathogenicity and Molecular Mechanisms
We utilized 13 prediction tools to evaluate the pathogenicity of BMPR1A p.R119C variant, including SIFT, PolyPhen-2, PROVEAN, P-Mut, Mutation_Assessor, CADD, REVEL, VEST4, FATHMM, GERP++, BayesDel, MPC, and PrimateAI. The SIFT algorithm evaluates how alterations in amino acid residues affect protein function and phenotype as an indicator of pathogenicity [24]. PolyPhen-2 allows physical comparison and potential categorization based on various sequence and structural features at substitution positions [25]. PROVEAN sequentially compares mutated protein to a reference sequence [26]. P-Mut could distinguish between neutral and disease-associated protein sequences [27]. Mutation_Assessor is based on evolutionary conservation of the affected amino acid in protein homologs [28]. CADD is a framework that combines various annotations to generate a single quantitative score [29]. REVEL is an ensemble method used to predict the pathogenicity of missense variants by utilizing individual tools [30]. VEST4 is a machine learning–based classifier designed to prioritize rare missense variants that may be associated with human disease [31]. FATHMM is a species-independent method that can predict the functional effects of protein missense variants, with the option of incorporating species-specific weightings [32]. GERP++ is a tool that utilizes maximum likelihood evolutionary rate estimation to provide position-specific scoring [33]. BayesDel is a versatile tool used for gene prioritization in gene discovery research and variant classification in clinical genetic testing [34]. PrimateAI aids in systematically identifying pathogenic mutations through a process of elimination [35]. MutPred2 is an independent sequence homology–based machine learning software that examines the structural, functional, and phenotypic outcomes of variants to forecast potential disease-causing molecular mechanisms [36]. Our predicted scores and pathogenicity results are based on the 2022 ClinGen calibration study [37].
2.4.2. Prediction of Structure and Binding Affinity
The protein structure of BMPR1A wild-type (WT) and the R119C mutant, including hydrogen bonds and disulfide bonds, was predicted using AlphaFold3 [38]. For each input, the model generated five predictions, and the predictions with the highest overall structure confidences were used for further analysis. All analyzed structures exhibited a predicted template modeling (pTM) score greater than 0.5, indicating that the overall predicted fold for the complex might be similar to the true structure [39].
The interaction structure of BMPR1A protein and BMP2 was predicted by AlphaFold3. Then, the predictions were visualized in ChimeraX for structural analysis including hydrogen bonds and contacts [40]. Additionally, PRODIGY, a software designed to predict binding affinity in biological complexes, was employed to further forecast protein–protein interactions [41].
2.4.3. Prediction of Protein Modifications and Alternative Splicing
Protein stability prediction was conducted using I-Mutant 2.0 [42], MUpro [43], FoldX [44], Rosetta [45], GeoDDG [46], DynaMut2 [47], DDMut [48], INPS-MD [49], DUET [50], mCSM [51], and SDM [52]. For FoldX, Rosetta, and GeoDDG, it is emphasized that negative DDG values indicate increased stability [53]; however, apart from them, all other prediction tools directly suggest that DDG < 0 indicates decreased stability [54].
For O-glycosylation prediction, we employed the NetOglyc 4.0 server to predict mucin-type O-glycosylation sites in mammalian proteins [55]; a score > 0.5 suggests positive effects. We also utilized updated tools including ISOGlyP and HOTGpred. ISOGlyP calculates the enhancement value product (EVP) as an indicator of glycosylation rate; an EVP value > 1 indicates a higher likelihood of glycosylation [56]. HOTGpred is a machine learning model for prediction, assigning a value to the probability of glycosylation for each protein [57]. For N-glycosylation prediction, we utilized the NetNglyc 1.0 server to identify potential N-glycosylation sites through artificial neural networks [58].
For ubiquitination and phosphorylation prediction, GPS-SUMO was applied to forecast sumoylation sites and SUMO interaction motifs in proteins [59]. Kinase prediction was conducted using NetPhos 3.1, a tool that employs an ensemble of neural networks to predict serine, threonine, or tyrosine phosphorylation sites in proteins [60].
Additionally, MusiteDeep is an online resource providing a deep learning framework for protein posttranslational modification (PTM) site prediction and visualization, including glycosylation, ubiquitination, and phosphorylation [61]. Sitetack is also an updated deep learning model that improves PTM prediction by using known PTMs [62].
For alternative splicing predictions, we employed the Human Splicing Finder (HSF) as a tool for predicting the impact of mutations on splicing signals or identifying splicing motifs in human sequence [63]. SpliceAI [64, 65] was also employed to predict splicing alterations. Its delta scores, ranging from 0 to 1, represent the probability that the variant influences splicing at any position.
3. Results
3.1. A Novel Pathogenic Missense Variant of BMPR1A in JPS
He was a 15-year-old male patient without any family history of cancer or polyposis, and his first colonoscopy showed that the entire colorectum is scattered with multiple polyps and there are more than 100 polyps in total; the largest is about 5∗5 cm in the rectum (Figure 1(a)). The patient has undergone multiple endoscopic polypectomy procedures, and most of the polyps were diagnosed pathologically as juvenile polyps (Figure 1(b)).
NGS showed that the patient carried a missense mutation in BMPR1A gene (c.355C>T; p.R119C), which is not included in multiple population databases (1000_CN, 1000_MAF, ESP6500, ExAC), and some literatures reported that this variant had been detected in families related to JPS [14]. This missense variant is located in the extracellular segment of the protein encoded by BMPR1A and is highly conserved in multiple species (Figure 1(c)). However, we performed 3D modeling analysis of the WT and mutant extracellular domains and found that the change of amino acid at Position 119 did not significantly affect the overall protein structures (Figure 1(d)) and the distribution of surrounding disulfide bonds (Figure 1(e)), albeit with a slight adjustment in hydrogen bonds (Figures 1(f) and 1(g)).
Next, in order to assess the pathogenicity of this missense variant, we employed various bioinformatics prediction tools to anticipate its potential functional implications. Positive results were obtained from 11 out of 13 widely used prediction tools (Table 1), indicating a high likelihood that the missense variant (p.R119C) might induce pathogenic alterations. Additionally, we utilized MutPred2, a predictive software for forecasting disease-causing molecular mechanisms triggered by SNP-induced amino acid substitutions. The results showed that BMPR1A p.R119C may be related to altered metal binding and altered transmembrane protein; furthermore, it may lead to gain of disulfide linkage at C124 and pyrrolidone carboxylic acid at Q117 (Table 1). These results present confident hypotheses for the possible pathogenicity as well as functional changes that may result from the mutation.
3.2. Prediction and Validation of the Interaction Between BMPR1A and BMP2
Previous studies have indicated that BMP2 interacts with BMPR1A and triggers the downstream TGF-β/BMP pathway [66]. Therefore, we utilized AlphaFold3 to model the structure of the BMPR1A-BMP2 interaction in order to predict whether BMPR1A p.R119C affects the interaction force between the proteins. The results showed that the mutation resulted in an increased protein binding affinity of BMPR1A with BMP2 (Table 2) and more interprotein hydrogen bonds and contacts (Figures 2(a) and 2(b)). It suggested that this missense variant might lead to aberrant overactivation of the TGF-β/BMP pathway. We then performed CoIP to verify protein interactions. Unfortunately, we did not observe satisfactory results for BMP2 binding. However, we thus discovered an interesting phenomenon that the variant (p.R119C) exhibited a lower molecular weight than the WT BMPR1A protein (Figure 2(c)). It has never been reported before, and we believe that it may also be a major cause of altered function. We then conducted further predictions and experimental analyses.
3.3. A Lower Molecular Weight in the BMPR1A p.R119C Variant
Changes in protein molecular weight are highly probably associated with PTMs, particularly glycosylation modifications. Therefore, we first carried out experiments to explore whether BMPR1A p.R119C was associated with altered glycosylation modifications. Results indicated that, after treatment with N-Gly or O-Gly, the missense variant may affect the N-glycosylation of the protein and have a modest effect on O-glycosylation (Figure 2(d)). Subsequently, we also performed the prediction of protein stability (Table 3) and various types of protein modifications (Table 4). The prediction results of protein stability indicated that 8 out of 11 prediction tools, including Rosetta, predicted a decrease in the stability of the mutated BMPR1A protein. In contrast, FoldX and GeoDDG showed a slight increase in stability. These findings suggest that the overall stability of the mutated protein may exhibit a slight decrease; however, there is still potential bias among different prediction results. Regrettably, no significantly plausible glycosylation alterations were identified across different prediction tools, apart from a minor change at one O-glycosylation site as observed within our experimental findings. However, through two updated deep learning frameworks for PTM prediction, namely, Sitetack and MusiteDeep, we found that its decreased phosphorylation level may also contribute to the decreased molecular weight of the protein.
Abnormalities in variable splicing may also bring about changes in molecular weight. Therefore, we utilized HSF and SpliceAI to further predict the potential splicing alterations induced by p.R119C (Table 5). The findings indicated that the mutation does not reside at the crucial splice site, thereby not significantly affecting the acceptor and donor sites; however, it may introduce a new exon splicing enhancer (ESE) site and disrupt two original exon splicing silencer (ESS) sites (Table 5). This implies that the mutation could also result in splicing disruption and potentially cause an early stop codon, thereby leading to a decrease in protein molecular weight. Thus, BMPR1A p.R119C may affect protein function through influencing variable splicing, as well as glycosylation and phosphorylation modifications.
4. Discussion
The BMPR1A p.R119C variant has been previously documented [14, 67]. A recent publication also proposed this missense variant in JPS with NGS and further demonstrated its potential pathogenicity through prediction tools like PolyPhen-2 [67]. However, they failed to offer a more in-depth analysis of the pathogenic mechanisms. In our case, this missense variant can be defined as pathogenic, according to the ACMG standard (Table 6).
The BMPR1A receptor is a crucial component protein of the TGF-β/BMP pathway, working in conjunction with other receptors such as BMPR1B and BMPR2, to synergistically receive signals from extracellular growth factors like BMP2, BMP4, and BMP6. This leads to pathway activation, with phosphorylation of SMAD1/5/8 molecules followed by complex formation with SMAD4 to regulate transcription of downstream target genes [68]. The BMP signaling pathway plays a vital role in stabilizing intestinal homeostasis and normal differentiation of the intestinal epithelium [69]. Abnormal disruption of the signaling pathway can largely impact normal proliferation of intestinal cells, as well as the tumor microenvironment balance [70]. Studies have shown that JPS development is closely associated with the BMP pathway. Sarah et al. have demonstrated that loss of BMPR1A-mediated signaling in fibroblasts could lead to the formation of serrated intestinal polyps [71]. However, its further specific pathogenic mechanisms remain unclear, including the precise impact of inactivation or overactivation of signaling pathways on the pathogenesis of JPS. In our CoIP results, we observed that the BMPR1A p.R119C variant did not affect BMP2 binding. Instead, we noted a decrease in the molecular weight. This implicated that the missense variant might disrupt the BMP pathway through other potential molecular mechanisms, ultimately resulting in JPS. This prompted us to conduct further investigation.
We are the first to propose that this BMPR1A missense variant may result in a decrease in protein molecular weight. As a prevalent PTM, glycosylation plays a crucial role in various biological processes such as protein stability, signal transduction, and cell–cell interactions [72, 73]. The formation of dysfunctional glycans is associated with disease states [74, 75]. Also, changes in glycosylation modifications can impact the three-dimensional structure and interaction affinity between the receptor and ligand [76]. Jonathan et al. have demonstrated that N-glycosylation on the BMPR2 receptor may specifically enhance BMP2 binding, which could mediate developmental and tissue homeostasis [77]. This provides substantial evidence for the potential impact of glycosylation modifications on protein function. As indicated in our findings, the variant might result in a reduction of protein N-glycosylation and slightly affect O-glycosylation. Our predictive results suggested that the variant might lead to a slight decreased protein stability, which aligns with the well-established role of glycosylation in maintaining protein conformation [78]. Although we did not observe a significant difference in subsequent glycosylation predictions, we still have grounds to argue that BMPR1A p.R119C is likely associated with an alteration in glycosylation modification, potentially leading to changes in protein conformation and molecular weight, ultimately impacting protein function.
Phosphorylation frequently induces conformational changes in proteins and plays a crucial regulatory role in various intracellular processes, including growth, proliferation, and cell division. Recent studies have elucidated the mechanisms by which aberrant phosphorylation contributes to cancer development [79–81]. Specifically, the gain or loss of phosphorylation sites has been identified as a crucial mechanism through which dysregulated phosphorylation impairs signal transduction [82]. Our results indicate that this mutated protein may exhibit reduced phosphorylation levels, which could influence its molecular weight. This finding further suggests that the R119C variant could lead to abnormalities in PTM processes, thereby affecting the normal function of this protein receptor and disrupting signal transduction.
Furthermore, we observed certain changes in mRNA splicing. Abnormal splicing may result in alternations within crucial functional protein domains, thus leading to their impaired functionality. Presently, numerous cancer-specific alternative splicing forms have been identified as pivotal factors influencing tumor progression and invasion [83], including colorectal malignancies [84]. A recent finding indicated that a newly identified BMPR1A mutation (c.778+5G>C) in JPS could influence mRNA splicing [85], suggesting that irregularities in alternative splicing might significantly contribute to the formation of intestinal polyps in JPS. However, they did not delve further into the specific effects of this variant on BMPR1A protein function. This prompted us to predict the splicing pattern for the R119C variant. While this particular locus does not affect the crucial splice site directly, we have a basis for arguing that it might disrupt regulators of alternative splicing based on the results of HSF. If the abnormal regulation results in frameshift and premature termination codons, then the observed reduction in protein molecular weight can be partially accounted for.
Overall, we have effectively demonstrated the pathogenicity of the BMPR1A R119C variant and comprehensively predicted its structural and functional alternations. Although our predictions and experimental investigations are still limited, the phenomenon of reduced protein molecular weight, initially proposed by us, could offer new insights for subsequent researchers to explore the pathogenic mechanism of BMPR1A protein in JPS.
5. Conclusion
In summary, we demonstrated that BMPR1A p.R119C is highly likely to be pathogenic. More importantly, we proposed for the first time that the missense variant would lead to a decrease in molecular weight, potentially associated with reduced protein stability, diminished PTMs, and aberrant alternative splicing. These findings may provide novel perspectives for further exploration into the role of BMPR1A in JPS development.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Howe J. R. Roth S. Ringold J. C. Mutations in the SMAD 4/DPC 4 Gene in juvenile polyposis Science 199828053661086108810.1126/science.280.5366.10862-s 2.0-00325240699582123 · doi ↗ · pubmed ↗
- 2Kay M. Eng K. Wyllie R. Colonic polyps and polyposis syndromes in pediatric patients Current Opinion in Pediatrics 201527563464110.1097/MOP.00000000000002652-s 2.0-8496008350826208235 · doi ↗ · pubmed ↗
- 3Syngal S. Brand R. E. Church J. M. ACG Clinical Guideline: Genetic Testing and Management of Hereditary Gastrointestinal Cancer Syndromes The American Journal of Gastroenterology 2015110222326210.1038/ajg.2014.4352-s 2.0-8496425224625645574 PMC 4695986 · doi ↗ · pubmed ↗
- 4Brosens L. A. A. Van Hattem A. Hylind L. M. Risk of colorectal cancer in juvenile polyposis Gut 200756796596710.1136/gut.2006.1169132-s 2.0-3434722159917303595 PMC 1994351 · doi ↗ · pubmed ↗
- 5Schreibman I. R. Baker M. Amos C. Mc Garrity T. J. The hamartomatous polyposis syndromes: a clinical and molecular review The American Journal of Gastroenterology 2005100247649010.1111/j.1572-0241.2005.40237.x 2-s 2.0-1394426564615667510 · doi ↗ · pubmed ↗
- 6Latchford A. R. Neale K. Phillips R. K. S. Clark S. K. Juvenile polyposis syndrome: a study of genotype, phenotype, and long-term outcome Diseases of the Colon & Rectum 201255101038104310.1097/DCR.0b 013e 31826278 b 32-s 2.0-8486870329622965402 · doi ↗ · pubmed ↗
- 7Papadopulos M. E. Plazzer J. P. Macrae F. A. Genotype–phenotype correlation of BMPR 1a disease causing variants in juvenile polyposis syndrome Hereditary Cancer in Clinical Practice 2023211 p. 1210.1186/s 13053-023-00255-337400896 PMC 10316536 · doi ↗ · pubmed ↗
- 8Langeveld D. van Hattem W. A. de Leng W. W. J. SMAD 4 immunohistochemistry reflects genetic status in juvenile polyposis syndrome Clinical Cancer Research 201016164126413410.1158/1078-0432.CCR-10-01682-s 2.0-7795573130520682711 PMC 2921472 · doi ↗ · pubmed ↗